Synthesis of Novel Heteropolycyclic Nitrogen Systems Bearing Fluorine Substituted Pyrazolo[3,4-d] Pyrimidine Derived from Polyfunctional π-Acceptor Compounds and Guanidine as Fungicidal Probes ()
1. Introduction
Recently, functionality substituted fluorinated pyrazolo[3,4-d] pyrimidine derivative, exhibited a wide spectrum in the biological active fields specially as enzymatic effects on cellobiase activity produced by some fungi [1] [2] [3] . Pyrazolopyrimidine derivatives possess a wide application of the biological activities, which encourage to research in this field, for example, antimicrobial [4] , antibacterial [5] , antitumor [6] , and anticancer [7] . In addition, introduction fluorine atoms to pyrazolopyrimidine enhance and improve their pharmacological properties [8] [9] . Abdel Rahman et al. [10] [11] [12] , reported that the orientation of cyclization reactions of functionalized amino and/or hydrazine bearing heterocyclic moieties depends on the effect of substituents, solvent pH, temperature, chemoselective orientation heterocycleization and regioselectivity of electrocyclization as well as preferring the cite of closure as this work focused on N’-heteroaryl guanidine 4 as electron donors towards various electron-acceptors reagents in view of their fungicidal effects.
2. Results and Discussion
Ortho diamines are active substrates for the building of a new heterocyclic nitrogen system [13] . In the case of unsymmetrical diamines, the substituents influence the initial participation of a particular amino group in the reaction, resulting in chemoselective products. In addition, the more electron withdrawing will be attacked firstly by primary amine [14] . Accordingly, the present work studies the interaction between N’-heteroaryl guanidine 4 with various poly-electron withdrawing centers in different media and conditions.
The starting material N’-heteroaryl guanidine 4 is obtained from condensation of 4-fluorobenzaledhyed with 4,5-dihydro-1-phenyl-3-methylpyrazol-5-one (1) in reflux EtOH/piperidine followed by cycloaddition via a nucleophilic attack with guanidine HCl under the same conditions to give 6-amino-4-(4’-fluoro-phenyl)-1-phenyl-3-methylpyrazolo[3,4-d]pyramidine (3) which upon a simple addition to cyanamide give N’-heteroaryl guanidine (4) (Scheme 1).
Interestingly the interaction between compound 4 with 4-nitrobenzoyl isothiocyanate (5) in non-polar solvent as dioxane led [12] give 1-heteroaryl-2-amino-6-aryl-1,3,5-triazin-4-thione (6) while some reaction in polar EtOH/piperidne, resulted in 1-(heteroaryl)-2-imino-4-aryl-1,3,5triazin-6 (SH) thione (7) (Scheme 2).
In addition, the interaction between guanidine derivative 4 and π-acceptors containing a carbonitrile group (8 and 10) in polar solvent as EtOH/Piperidne the more electronegativity cite will attacked firstly before a moderate electronegativity cite via electrocyclization reaction [12] to give 1-(heteroaryl)-2-imino-4-amino-6-aryl-pyrim-idine-5-yl-carbonitrile (9) and/or 1-(heteroaryl)-2-
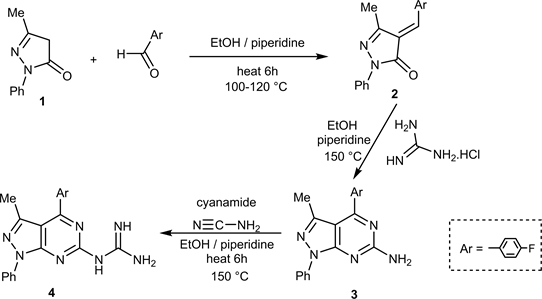
Scheme 1. Synthesis of compounds 2, 3, and 4.
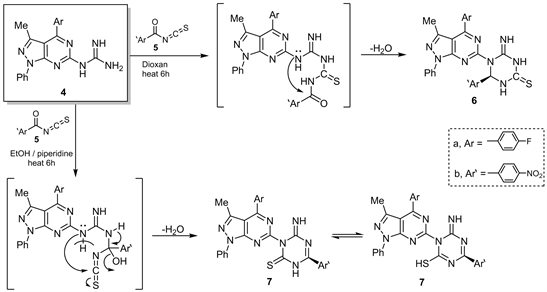
Scheme 2. Synthesis of compounds 6 and 7.
imino-4-amino-6-aryl-pyrimidine-5-carboxylic acid (11) respectively (Scheme 3). Compound 11 gave the acidity test with aqueous NaHCO3.
Cycloaddition reaction of N’-heteroaryl guanidine 4 with 1-phenyl-3-methyl-4-arylidene-pyrazol-5-one (12) in reflux EtOH/piperidine led to the direct formation of pyrazolopyrimidine derivative 13 via a nucleophilic attack (Scheme 4).
Finally, the introduction of F-atoms to heterocyclic nitrogen systems often improve their physical, chemical and biological properties [15] [16] . Thus, cyclocondensation of N’-(heteroaryl) guanidine 4 with fluorinated 1,3-bicarbonyl compounds as hexafluoro acetylacetone (14) in reflux in EtOH, afforded N’-(heteroaryl)-2-imino-4,6 di(trifluoromethyl)pyrimidine (15) (Scheme 5).
As recently, the synthesis of pyrazolopyrimidine moiety bearing other heterocyclic systems as bioactive semidrugs was reported through a type of nucleophilic attack toward a move positive electrophilic center followed by cycloaddition reaction [1] [2] [17] [18] [19] [20] , this investigation was focused on the synthesis of novel fluorine substituted heteropolycyclic nitrogen systems containing a pyrazolopyrimidine moiety in view of their cellobiase activity towards some fungi.
The structure of the products were deduced from correct elemental analysis and their spectral measurements. The reagents used prepared according to Abdel-Rahman et al. [13] [14] .
IR absorption spectral study of the obtained systems 6, 7, 9, 11, 13, and 15 give us a good indication about their structural.
IR spectra of compound 6 recorded γ at 3200 of NH2, while that of 7 showed both =NH and NH at γ 3362 and 3160 cm−1. Compounds 6 and 7 showed γ at 3038, 3060 for aromatic CH and γ at 2927, 2286 for aliphatic CH and γ at 1580, 1581 for C=N, and γ at 1341, 1321 for symmetric and asymmetric NO2, and γ at 782, 746 for C-F bands. Also compound 6 showed γ at 1128 for C=S.
Additionally, the IR spectrum of both compounds 9 and 11 was recorded γ at 2220 for CN (9) and γ at 3363, 1694 cm−1 for OH and C=O functional groups
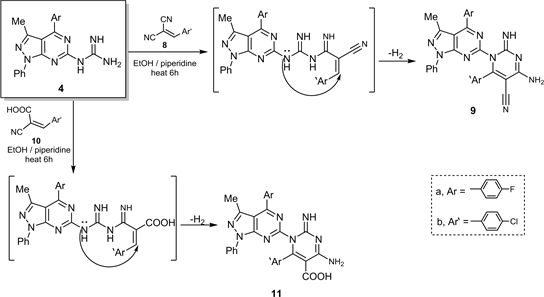
Scheme 3. Synthesis of compounds 9 and 11.
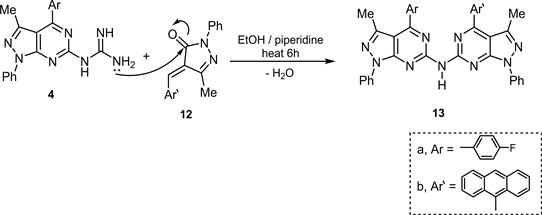
Scheme 4. Synthesis of compound 13.
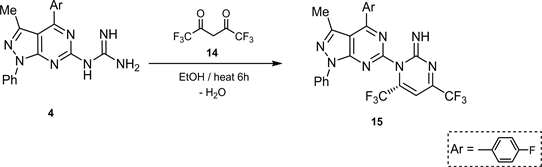
Scheme 5. Synthesis of compound 15.
(11) with the presence the functional of =NH, NH2, C-F, while compound 13 showed only =NH at γ 3227 cm−1 with lacks of both C=O, NH2 and C=C, which confirm the cycloaddition reaction. As expected, Compound 15 showed γ 3320 of =NH with lacks of both NH2 and C=O functional groups. Similarly, all the synthesized compounds showed γ characterized for aryl aliphatic, C=N, and C-F functional groups.
Furthermore, 1H NMR spectrum of compound 6 showed a resonated signal at δ 3.5 ppm for NH2, while that of 7 recorded at δ 10.55 and 8.5 ppm for =NH and NH proton compounds 9 and 11 attributed to =NH and NH protons, showed slight changed recordings at δ 10.55 and 11 ppm for =NH and showed at δ 3.5 and 3.25 ppm respectively for NH2, although 11 exhibit a resonated signal at δ 9.5 ppm for OH.
On the other hand, 1H NMR spectra of compounds 13 and 15 showed only one =NH protons at approximately 11.0 and 10.50 ppm.
Focused on function group 13C NMR spectra of compounds 6 and 7 showed at δ 182 - 185 ppm respectively for C=S carbons, while that of 9 and 11 exhibits δ at 150 and 162 ppm for CN and C=O.
All the 13CNMR spectra showed also, δ at 145 - 143, 140, 132 - 122 and 35 - 30 ppm for C-F, C=N, aryl and methyl carbons.
Finally, mass spectral study of some isolated heterobicyclic nitrogen systems reported a M+ with the two major bulky fragments which undergo farther fragmentation gave the selected base peak at m/e 95 as 4-fluorophenyl ion (11 and 15) (Figure 1 and Figure 2).
![]()
Figure 1. Mass fragmentation pattern of compound 11.
![]()
Figure 2. Mass fragmentation pattern of compound 15.
3. Antifungal Activity (Cellobiase)
Due to the medicinal, pharmacological and biological properties of fluorine bearing fused heterobicyclic nitrogen systems the present word tends to evaluate the new fluorine substituted heterobicyclic nitrogen systems as enzymatic affects on the cellobiase produced by fungi [21] [22] [23] . The in vitro antifungal activity of the new fluorinated systems obtained via inhibition of mycelial growth of Penicillium italicum, Helimenthosporiumsatium, Pythium deberyanum and Fusarium solani in methanol and sterile potato dextrose agar (PDA) [23] .
The fungi toxic activity of the synthesis compounds are tested on P. italium. A Discs of orange rinds (3 × 3 cm) were removed from orange fruits. The discs were further sterilized by 70% ethanol, the rinds were treated with the tested compounds. The treated discs were allowed to dry and were artificially inoculated with spots of P. Itlaicum. The Commercial thiobendazol-2-(4-thiazolyl) benzimidazole (TBZ) Figure 3 was used as control.
The result percentage of rotted discs were evaluated after one weeks Table 1.
Prevention of blue mold development the action of new fluorinated systems obtained on the decay control on rind discs is present in Table 2. The results indicate that only compound 15 gave good result in comparison with control. On the other hand, compounds which had the same higher ED50 values did not prevent the decay at all concentration used.
![]()
Table 1. Antifungal activity of new fluorinated systems toxicity index.
aED50 for inhibition of mycellal growth expressed.
![]()
Table 2. Effect of new fluorinated pyrimidine on prevention of disease development on rinddiscs.
![]()
Figure 3. 2-(4'-Thiazolyl) benzimidazole.
According to the Table 1 and Table 2 we suggest the following conclusion:
• The antifungal activity of tested compounds based on ED50 are 15 > 11 > 9 > 6, 7 > 13.
• Compound 15 gave a good activity in comparison with control TEZ especially at 4000 µg∙ml−1 may be due to the higher effect of CF3 groups on the biologically sensitivity of tested fungi.
• The compounds which had the same or higher ED50 values did not prevent the decay at all the concentrations used.
4. Conclusion
In conclusion, this study provides a short and reasonable low cost route to performance N-substituted guanidine towards some α, β, γ-polyfunctional reagents in different conditions to contribute fused heteropolycyclic nitrogen systems as antifungal probes. These compounds have been tasted as antifungal activity, which showed the compounds had the same higher ED50 values did not prevent the decay at all concentration used. The compound 15 has a rich F-atoms exhibited a highly antifungal activity in comparison with control Thiobendazole (TBZ) which can be effective on fungal disease in orange tree.
5. Experimental
Melting points determined by an electrochermal Bibly Sturat Scientific melting point sample (UK). A Perkin Elmer Model RXI-FT IR system 55,529 used for IR spectra of the prepared compounds (cm−1). A Bruker advance DPX 400 MHZ model uses TMS as internal standard was used for recording the 1H and 13C NMR spectra of the compounds on deuterated DMSO-d6 (ppm). Elemental analysis was performed in micro analytical Center of Cairo University, Cairo, Egypt.
The common organic reagents 5, 8, 10 and 12 were obtained according to the reported methods [11] [12] . Only compound 5 produced from reflux of ammonium thiocyanate with 4-nitrobenzoyl chloride in dry acetone 4-(4’-Fluorobenzylidene)-1-phenyl-3-methyl-pyrazol-5-one (2) and 6-amaino-4-(4’-Fluorophenyl)-1-phenyl-3-methyl-pyrazolo[3,4-d]pyrimidine (3) were obtained according the reported method [17] .
1-(1’-phenyl-3’-methyl-4’-(4"-fluorophenyl)-pyrazolo [3,4-d] pyrimidine-6’-yl)-2-amino-6-(4’-nitrophenyl)-1,3,5-triazin-4-thione (6).
A mixture of 4 (0.01 mol) and 5 (0.01 mol) in dry dioxane (100 ml) refluxed for 6 h, cooled. The solid obtained filtered off and crystallized from dioxane to give 6 as yellowish crystals.
Yield 68% m.p. > 350 ˚C. IR (γ) cm−1: 3200 (NH2), 3038 (aromatic CH). 2927, 2888 (aliphatic CH), 1612 (C=C), 1580 (C=N), 1341 (NO2), 1474, 1457 (bending CH3), 1158 (C=S), 1211 (C-F), 861, 820 (substituted aryl), 782 (C-F). 1H NMR (DMSO-d6) δ ppm: 8.4, 8.2 and 8.0, 7.8 (each dd, CH-F and CH-NO2), 7.6-7.35, 7.2 - 6.91 (each m, 9H, aromatic CH), 3.55 (s, NH2), 1.50 (s, 3H, CH3). 13C NMR (DMSO-d6) δ ppm: 182 (C=S), 145 (C-F), 142 (C=N), 138 (C-NO2), 130 - 122 (aromatic carbons), 38 (C-CH3 carbon). Aral. Calcd.; C, 58.58%; H, 3.64%; F, 3.43%; N, 22.77%; S, 5.79% for C27H18N9FSO2 (553). Found: C, 58.49%; H, 3.22%; F, 3.21%; N, 22.61%; S, 5.50%.
1-(1’-phenyl-3’-methyl-4’-fluorophenyl-pyrazolo[3,4-d]pyrimidine-6’-yl)-2-imino -4-(4’-nitrophenyl)-1,3,5-triazin-6 (SH) thione (7).
Equimolar mixture of 4 and ‘5 in ethanol (100 ml) with drops of piperidone refluxed for 6 h, cooled then adds drops of acetic acid. The produced solid, filtered off and crystalized from dioxane to give 7 as yellowish crystals.
Yield 75%, m.p. > 340˚C. IR (γ) cm−1: 3362 (=NH), 3160 (NH), 3060 (aromatic CH), 2886 (aliphatic CH), 1603 (C=C), 1581 (C=N), 1321 (NO2), 1271 (C-F), 879, 849 (substituted phenyl), 746 (C-F). 1H NMR (DMSO-d6) δ ppm: 10 - 55 (=NH), 8.5 (NH), 8.4, 8.25, 8.0, 7.95 (each d.d CH-F and CH-NO2). 7.80 - 7.66, 7.45 - 6.95 (each m, 9H, aromatic CH), 1.2 (s, CH3). 13C NMR (DMSO-d6) δ ppm: 185 (C=S), 148 (C-F), 146 (C=N), 140 (C-N), 135 (C-NO2), 130 - 124 (aromatic carbons), 34 (C-CH3). Aral. Calcd.; C, 58.80%; H, 3.29%; F, 3.44%; N, 22.86%; S, 5.81% for C27H18N9FSO2 (551). Found: C, 58.65%; H, 3.08%; F, 3.19%; N, 22.51%; S, 5.55%.
1-(1’-phenyl-3’-methyl-4’-(4"-fluorophenyl)-pyrazolo[3,4-d]pyrimidine-6’-yl)-2-imino-4-amino-6-(4’-chlorophenyl)-pyrimidine-5-yl-carbonitrile (9).
A mixture of 4 (0.01 mol) and 8 (0.01 mol) in EtOH (100 ml) with drops of piperidine refluxed for 6 h, cooled then poured onto ice. The solid resulted, filtered off and crystalized from EtOH to give 9 as deep brown crystals.
Yield 65%, m.p. > 330 ˚C. IR (γ) cm−1: 3350 (=NH), 3150 (NH2), 2220 (CΞH), 1607 (C=C), 1449 (bending CH3), 1254 (C-F), 968, 880, 798 (substituted phenyl), 752 (C-F), 650 (C-Cl). 1H NMR (DMSO-d6) δ ppm: 10.55 (s, =NH), 8.2, 8.0, 7.8, 7.75 (each d.d CH-F and CH-Cl), 7.66 - 7.40, 7.2 - 6.85 (each m, 9H, aromatic CH), 3.5 (s, 2H, NH2), 1.25 (s, 3H, Me). 13C NMR (DMSO-d6) δ ppm: 150 (CN), 147 (C-F), 142 (C=N), 140 (C-Cl), 132-121 (aromatic carbons), 35 (C-CH3 carbon). Aral. Calcd.; C, 63.56%; H, 3.50%; Cl, 6.47%; F, 3.47%; N, 23.00% for C29H19N9FCl (547). Found: C, 63.41%; H, 3.29%; Cl, 6.25%; F, 3.31%; N, 22.89%.
1-(1’-phenyl-3’-methyl-4’-(4"-fluorophenyl)-pyrazolo[3,4-d] pyrimidine-6’-yl)-2-imino-4-amino-6-(4’-chlorophenyl)-pyrimidine-5-carboxylic acid (11).
A mixture of 4 (0.01 mol) and 10 (0.01 mol) in EtOH (100 ml) with drops of piperidine refluxed for 6 h, cooled then poured onto ice. The solid produced filtered off and crystalized from dioxane to give 11 as deep brown crystals.
Yield 60%, m.p. 198˚C - 200˚C. IR (γ) cm−1: 3363 (OH), 3300 (=NH), 3150 (NH2), 2888 (aliphatic CH3), 1694 (C=O), 1609 (C=C), 1585 (C=N), 1490 (bending CH3), 1272 (C-F), 940, 879, 859 (substituted phenyl), 746 (C-F), 609 (C-Cl). 1H NMR (DMSO-d6) δ ppm: 11 (s, =NH), 9.5 (s, 1H, OH), 8,9 (s, 1H, NH), 8.4, 8.2 and 7.9, 7.7 (each d.d CH-F and CH-Cl), 7.5 - 7.25, and 7.1 - 6.8 (each m, 9H, aromatic CH), 3.25 (s, 2H, NH2), 1.25 (s, 3H, Me). 13C NMR (DMSO-d6) δ ppm: 162 (C=O), 145 (C-F), 142 (C=N), 139 (C-NO2), 131 - 123 (aromatic carbons), 39 (C-CH3 carbon). M/S (m/e, Int. %) = 568 (M + 2, 1.1), 303 (2.15), 264 (1.18), 138 (65.11), 132 (15.13), 111 (25.0), 95 (100), 68 (13.13). Aral. Calcd.; C, 61.43%; H, 3.56%; Cl, 6.25%; F, 3.35%; N, 19.76% for C29H2ON8FClO2 (566). Found: C, 61.31%; H, 3.41%; Cl, 5.91%; F, 3.15%; N, 19.59%.
1-(1’-phenyl-3’-methyl-4’-(4"-fluorophenyl)-pyrazolo[3,4-d]pyrimidine-6’-yl)-2-imino-4-methyl-6-phenyl-7-(anthracen-9’-yl)-pyrimido[5,4-d]pyrazole (13).
Equimolar mixture of 4 and 12 in EtOH (100 ml) with drops of piperidine refluxed for 6 h, cooled then poured onto ice. The yielded solid, filtered off and crystalized from dioxane to give 13 as deep violet crystals.
Yield 72%, m.p. 170˚C - 172˚C. IR (γ) cm−1: 3227 (=NH), 3060 (aromatic CH), 2880 (aliphatic CH), 1609 (C=C), 1250 (C=N), 1453 (bending CH3), 1224 (C-F), 908, 850, 800 (substituted phenyl), 752 (C-F), 600 (C-Cl). 1H NMR (DMSO-d6) δ ppm: 11 (s, 1H, =NH), 8.4, 8.11 and 8.0, 7.88 (each d.d CH-F and C2H and C8H of anthracene), 7.7 - 7.41, 7.2 - 6.9, 6.6 - 6.4 (each m, 18H, aromatic protons). 1.45 and 1.25 (each s, 2 CH3, protons). 13C NMR (DMSO-d6) δ ppm: 145 (C-F), 142 (C=N), 138 (C=C), 132 - 124 (aromatic CH), 40 and 36 (2 CH3 carbon). Aral. Calcd.; C, 75.09%; H, 4.30%; F, 2.70%; N, 17.91% for C44H30N9F (703). Found: C, 74.91%; H, 4.11%; F, 2.22%; N, 17.75%.
1-(1’-phenyl-3’-methyl-4’-(4’-fluorophenyl)-pyrazolo[3,4-d]pyrimidine-6’-yl)-2-imino-4,6-di(trifluoromethyl) pyrimidine (15).
A mixture of 4 (0.01 mol) and 14 (0.01 mol) in EtOH (50 ml) with drops of piperidine refluxed for 2 h, cooled then poured onto ice. The yielded solid, filtered off and crystalized from MeOH to give 15 as yellowish crystals.
Yield 78%, m.p. > 350˚C. IR (γ) cm−1: 3320 (=NH), 3039 (aromatic CH), 2887 (aliphatic CH), 1612 (C=C), 1598 (C=N), 1474, 1447 (bending CH3), 1211 (C-F), 907, 861, 822, 783 (substituted phenyl), 750 (C-F), 721 (C-F), 695 (C-F). 1H NMR (DMSO-d6) δ ppm: 10.5 (s, 1H, =NH), 8.9 (d. 1H, C5-H of hexafluoromethyl pyrimidine), 8.2, 8.0 (d. CH-F), 7.7 - 7.4, 7.2 - 6.88 (each m, 9 H, aromatic protons), 1.25 (s, 3H, CH3). 13C NMR (DMSO-d6) δ ppm: 148 (=NH), 144 (C-F), 140 (C-N), 130 - 124 (aromatic CH), 40 (C-CH3 carbon). M/S (m/e, Int. %) = 535 (M + 2, 5.11), 303 (55.18), 230 (17.15), 170 (85.01), 122 (23.0), 108 (8.90), 95 (100). Aral. Calcd.; C, 54.04%; H, 2.65%; F, 24.93%; N, 18.38% for C24H14N7F7 (533). Found: C, 53.89%; H, 2.41%; F, 24.65%; N, 18.59%.
Acknowledgements
The authors would like to thank the Chemistry Department at King Abdulaziz University, Jeddah, for supporting this Research. Thanks to Prof Reda Abdel-Rahman (D.Sc. Organic chemistry), for his help and constant support.