Targeting HIF-1α and HIF-2α to Overcome Treatment Resistance Mediated by Oncogenic KRAS in Colorectal Cancer ()
1. Introduction
The epidermal growth factor receptor (EGFR) is a transmembrane tyrosine kinase receptor that is frequently overexpressed in epithelial cancers. Signaling pathways downstream of EGFR have been identified; which include the Ras/Raf mitogen-activated protein kinase (MAPK) pathway, the phosphoinositide-3 kinase (PI3K)/AKT pathway, and the Jak2/STAT3 pathway. Activation of these parallel signaling pathways then leads to the induction of HIF-1α and HIF-2α, and the consequent transactivation of target genes [1-4] . Blockade of EGFR, through the regulation of these signaling pathways, would inhibit tumorigenesis and lead to improved responses to chemotherapy and radiation therapy.
Two strategies to inhibit EGFR have been developed for clinical use [5-7] . Cetuximab and panitumumab are monoclonal antibodies that target EGFR and function by binding to its extracellular domain with high affinity, blocking its ligand from binding. Erlotinib, gefitinib, and lapatinib are small-molecule tyrosine kinase inhibitors (TKI) that function by inhibiting EGFR autophosphorylation and downstream signaling. Although targeting EGFR has shown promise, clinical efficacy has been modest; and even in those patients who show a response, benefits are short-lived. One of the major resistance mechanisms is activating mutations in oncogenic KRAS; which leads to primary resistance to EGFR inhibitors.
2. Effect of KRAS Mutational Status on Treatment with EGFR Inhibitors in Colorectal Cancer (CRC) (Table 1)
Studies assessing the efficacy of anti-EGFR monoclonal antibodies (mAb) in colorectal cancers have suggested that their efficacy is confined to patients with wild-type KRAS. In this article, we will summarize results from these trials.
Tol et al. evaluated the effect of adding cetuximab to the combination of capecitabine, oxaliplatin, and bevacizumab for previously untreated metastatic colorectal cancer [8] . The mutational status of the KRAS gene was evaluated in 528 tumors. An activating KRAS mutation was found in 206 tumors (39.6%)—108 from patients in the cetuximab-treated arm and 98 from patients in the

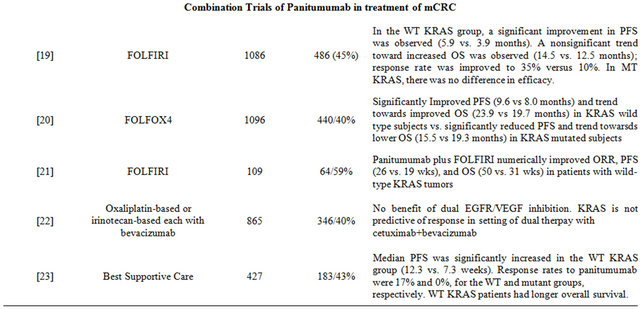
Table 1. Clinical trials with EGFR inhibitors.
control group. Although the addition of cetuximab resulted in similar objective response rate (ORR) and overall survival (OS), it also lead to a statistically significant shorter progression-free survival (PFS) and inferior quality of life score. In addition, cetuximab-treated patients with mutated KRAS had significantly shorter PFS than cetuximab-treated patients with wild-type KRAS tumors or patients with mutated KRAS tumors in the control group.
Bokemeyer et al. corroborated these findings when they compared folinic acid, 5-FU and oxaliplatin (FOLF OX-4) plus cetuximab to FOLFOX-4 alone [9] . In their updated phase II analysis of OPUS (Oxaliplatin and Cetuximab in First-Line Treatment of Metastatic Colorectal Cancer) published in 2011, KRAS status was available for 315 patient samples (93% compared to 69% in original report] [10] . Fifty-seven percent of these tumors had wild-type KRAS. In this subset of patients, the addition of cetuximab to FOLFOX-4 significantly improved PFS and ORR. On the other hand, patients whose tumors carried mutations in KRAS and received cetuximab plus FOLFOX-4, odds of response were lower and risk of disease progression were higher when compared to the FOLFOX-4 alone arm.
In one of the largest analyses to date, De Roock and colleagues gathered 1022 tumor DNA samples from patients treated with cetuximab between 2001 and 2008 from 11 centers in seven European countries [11] . Mutational analysis for KRAS as well as other downstream mutations was performed. The analysis found that patients with KRAS mutation did not derive benefit compared with patients with wild-type KRAS, with a response rate of 6.7% versus 35.8%, a median PFS of 12 weeks versus 24 weeks, and a median OS of 32 weeks versus 50 weeks. The relationship of BRAF and NRAS mutations to KRAS was also evaluated. In KRAS wildtypes, carriers of BRAF and NRAS mutations had a significantly lower response rate than did BRAF and NRAS wild-types. Multivariate analysis and conditional inference trees confirmed that, if KRAS is not mutated, assessing BRAF, NRAS, and PIK3CA mutations gives additional information about outcome. ORRs were 24.4% in the unselected population, 36.3% in the KRAS wildtype population, and 41.2% in the KRAS, BRAF, NRAS, and PIK3CA wild-type population.
Van Custem et al. reported results of a randomized controlled trial of cetuximab plus folinic acid, 5-FU and irinotican (FOLFIRI) as first line therapy for mCRC in 2009 [12] . They also found that the benefit of cetuximab was limited to KRAS wild-type tumors. They published updated results in 2011 after ascertaining KRAS mutation status in 89% of samples confirming their initial results [13] . They found significant interactions between KRAS status and all efficacy endpoints—PFS, ORR and OS. BRAF tumor mutation was also found to be a strong indicator of poor prognosis.
In a phase II study to evaluate the efficacy and safety of cetuximab along with FOLFOX, Colucci et al. performed mutational analysis of KRAS and BRAF genes in 35 of the 69 patients treated with cetuximab (51%) [14] . KRAS was mutated in 13 out of the 35 cases (37%), whereas no mutations were detected in the BRAF gene. A trend toward an association between KRAS mutations and objective response (OR) to treatment was demonstrated. Analysis of survival showed that patients harboring KRAS mutations had a trend toward shorter time to progression (TTP). Indeed, KRAS mutations were significantly associated with a poorer OS in both unadjusted analysis and ageand sex-adjusted Cox multivariate regression.
Folprecht et al. studied the efficacy of cetuximab in combination with 5-FU based therapy in mCRC patients with unresectable liver metastases [15] . The authors also performed a retrospective analysis of response by KRAS status. A partial or complete response was noted in 47 of 67 (70%) patients with KRAS wild-type tumors versus 11 of 27 (41%) patients with KRAS-mutated tumors. Resectability rates increased from 32% (22 of 68 patients) at baseline to 60% (41 of 68 patients) after chemotherapy. This study provides a new dimension to the use of KRAS as a biomarker in presence of liver metastases.
In the cetuximab plus FOLFOX-6 or FOLFIRI in metastatic colorectal cancer trial, Ocvirk et al. evaluated the efficacy and safety of cetuximab combined with FOL FOX (n = 74) or FOLFIRI (n = 77) [16] . Although no significant difference in PFS, ORR, or median OS could be detected between the treatment arms, KRAS mutation status (determined in a subset of 117 patients) was found to be predictive of response. Patients with KRAS wildtype tumors demonstrated improved PFS, OS, and ORR.
It is worth noting that all the above trials reported data on KRAS mutation status retrospectively and were not designed to assess a difference in efficacy of cetuximab by KRAS mutation status. More recently, Maughan et al. published the results of their Medical Research Council (MRC) COIN trial [17] . In this large prospective clinical trial of 1630 patients, cetuximab was added to standard chemotherapy in first-line treatment of advanced colorectal cancer with the primary aim of assessing its effect on OS in patients with wild-type KRAS tumors. Overall survival and PFS did not change with the addition of cetuximab to standard therapy. Overall response rate increased from 57% (n = 209) with chemotherapy alone to 64% (n = 232) with addition of cetuximab (p = 0.049). However, somatic mutations remained predictive of OS irrespective of treatment received: BRAF mutant, 8.8 months; KRAS mutant, 14.4 months; wild-type for both BRAF and KRAS, 20.1 months.
Another prospective randomized phase II trial investigated the efficacy and safety of cetuximab combined with capecitabine and irinotecan (CAPIRI) or capecitabine and oxaliplatin (CAPOX) in the first-line treatment of mCRC [18] . Of the 177 randomized patients, analysis of the KRAS gene mutation status was performed in 81.4% of the intention to treat population. The ORR and PFS were comparable in the KRAS wild-type and mutant subgroups; however a trend toward longer survival was associated with KRAS wild-type.
Peeters et al. evaluated the efficacy and safety of panitumumab plus FOLFIRI compared with FOLFIRI alone as a second line therapy after failure of initial treatment for mCRC in a prospective randomized phase III trial [19] . One hundred and eighty-six patients were randomly assigned to one of these treatment arms on a 1:1 ratio and the results were evaluated by KRAS status (available for 91% of patients). In the wild-type KRAS subpopulation, a significant improvement in PFS (5.9 months vs. 3.9 months) was observed. A trend toward increased OS was also observed (14.5 months versus 12.5 months) and response rate was improved to 35% versus 10%. However there was no difference in efficacy in patients with mutated KRAS.
In the Panitumumab Randomized Trial in Combination with Chemotherapy for Metastatic Colorectal Cancer to Determine Efficacy (PRIME) study, a similar study design was used [20] . Douillard et al. designed this trial to evaluate the efficacy and safety of panitumumab plus FOLFOX-4 versus FOLFOX-4 alone as initial treatment for mCRC. Results were analyzed by KRAS status (available for 93% of the 1183 patients). In the wild-type KRAS subpopulation, addition of panitumumab to FOLF OX-4 significantly improved median PFS (9.6 months vs. 8.0 months). A nonsignificant increase in OS was also observed for panitumumab plus FOLFOX-4 group (median OS, 23.9 months vs. 19.7 months). In the mutant KRAS subpopulation, PFS was significantly reduced by the addition of panitumumab and a trend towards lower OS was observed. This study again demonstrated the efficacy of panitumumab in patients with wild-type KRAS tumors and not those harboring a mutation in KRAS.
In an open-label, single-arm phase II trial, Cohn et al. prospectively evaluated the effect of tumor KRAS status on efficacy of panitumumab plus FOLFIRI as second line treatment in patients with unresectable, measurable mCRC after failure of first-line treatment with oxaliplatin-based chemotherapy plus bevacizumab [21] . Of 116 patients enrolled, 109 patients with known tumor KRAS status received treatment; 59% had wild-type KRAS, and 41% had mutant KRAS. Fifteen patients (23%) with wild-type KRAS and 7 patients (16%) with mutant KRAS had a complete or partial response to treatment. Median PFS was 26 weeks and 19 weeks, and median OS was 50 weeks and 31 weeks in the wild-type KRAS and mutant KRAS groups, respectively. Thus, panitumumab plus FOLFIRI numerically improved ORR, PFS, and OS in favor of patients with wild-type KRAS tumors.
Hecht et al. evaluated the association of tumor EGFR expression levels with outcomes in patients with chemotherapy-refractory mCRC in a phase II, multicenter, single-arm, open-label study [22] . KRAS mutational status was also evaluated in this study. A total of 203 patients classified as low/negative EGFR (1% - 9%) and 185 patients with high EGFR (> or = 10%) were enrolled in the studies. Overall response rate was 5.7% in patients with low/negative EGFR and 4.2% in patients with high EGFR; response rate at week 16 was 4% in both groups (all partial responses). Median PFS were 8.1 weeks, 8.1 weeks, and 7.3 weeks in patients with negative, low, and high levels of EGFR expression, respectively. Although EGFR expression was not predictive of outcome, PFS and OS were longer in patients with wild-type KRAS in comparison to those with mutant KRAS.
Panitumumab monotherapy was compared to best supportive care in a phase III trial in patients with chemotherapy-refractory mCRC [23] . KRAS status was ascertained in 427 (92%) of 463 patients (208 panitumumab, 219 best supportive care). KRAS mutations were found in 43% of patients. Treatment effect on PFS and OS in the wild-type KRAS group (12.3 weeks and 6.8 months) was significantly greater than in the mutant group (7.3 weeks and 4.5 months). Response rates to panitumumab were 17% and 0% for the wild-type and mutant KRAS groups, respectively.
3. Targeting HIF-1α and HIF-2α to Overcome Treatment Resistance Due to KRAS Mutation
Oncogenic RAS mutations are found in approximately 30% of all human tumors, with KRAS being the most prevalent. KRAS mutations are most prevalent in pancreatic (72% - 90%), thyroid (55%], colorectal (32% - 57%), and lung cancers (15% - 50%) [24]. Point mutations at codons 12, 13, or 61 result in stabilization of KRAS in the GTP-bound conformation, rendering it constitutively active. Activated RAS oncoproteins have been shown to activate MAPK, PI3K/AKT, and Jak2/STAT3 signaling; and thereby induce expression and transcripttional activity of the hypoxia-inducible factors-1α and -2α (HIF-1α and HIF-2α) [25-27] .
HIF-1α and HIF-2α are transcription factors that are overexpressed in cancer and linked to cancer progression. Structurally, HIF-1α and HIF-2α are partially related, sharing 48% overall amino acid identity and two identical proline residues in their oxygen-dependent degradation domains. HIF-1α and HIF-2α dimerize with HIF-1β to form HIF-1 and HIF-2, respectively. Overexpression of these heterodimers is driven by intratumoral hypoxia. Under normoxia, HIF-1α and HIF-2α are ubiquitinated through an oxygen-dependent interaction with the von Hippel-Lindau protein (pVHL) and degraded by the 26S proteasome. Under hypoxic conditions, HIF-1α and HIF-2α proteins accumulate, translocate to the nucleus, dimerize with HIF-1α, and transactivate target genes.
In cancer, intratumoral hypoxia and genetic alterations in tumor suppressor genes and oncogenes induce HIF-1α and HIF-2α overexpression. MAPK, PI3K/AKT, and Jak2/STAT3 signaling pathways downstream of oncogenic RAS also phosphorylate HIF-1α and, thereby, stimulate its transcriptional activity. HIF-1α and HIF-2α work together to regulate genes signature overlapping with oncogenic KRAS [28]. Both of these proteins are overexpressed in many cancer types; and their overexpression is correlated with adverse outcome [29,30] . HIF-1α and HIF-2α bind to hypoxia response elements (HRE) on the promoters of target genes, and transactivate both unique and overlapping sets of genes, which then cause cancer cell proliferation, resistance to apoptosis, neo-angiogenesis, invasion, and metastasis.
Due to their roles in integrating signaling downstream of oncogenic KRAS, HIF-1α and HIF-2α would serve as an Achilles’ heel to target for cancer therapy. To date, high-throughput small-compound screens and mechanistic studies have identified several classes of anticancer agents that disrupt HIF-α function, including inhibition of its transcriptional activity and HIF-α protein synthesis or stability [31-34] . Many of these HIF inhibitors are currently in phase I and II clinical trials (Table 2 and Figure 1). Future clinical development to find inhibitors targeting KRAS-mutated cancers should include trials with HIF inhibitors. Appropriate doses and sequencing of these agents in combination with standard therapy would

Table 2. HIF inhibitors in clinical development.
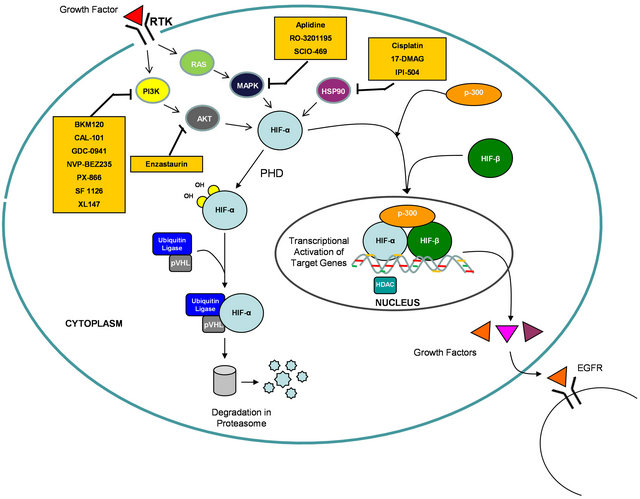
Figure 1. Diagram depicting inhibitors of the HIF pathway in clinical development.
need to be tested in preclinical models to help with clinical trial design.
NOTES
#These authors contributed equally.
†Corresponding authors.