Diabetes, an inflammatory process: Oxidative Stress and TNF-alpha involved in hepatic complication ()
1. INTRODUCTION
After food intake, blood glucose levels rise and insulin is released by the pancreas to maintain homeostasis. In the diabetic state, the absence or deficient action of insulin in target tissues is the cause of hyperglycemia and abnormalities in the metabolism of proteins, fats and carbohydrates. In addition, chronic hyperglycemia, a hallmark of diabetes, is responsible for organic dysfunction, being eyes, kidneys, nervous system, heart and blood vessels the most important organs affected. Diabetes mellitus (DM) is a heterogeneous deregulation of carbohydrate metabolism, characterized by chronic hyperglycemia that results from impaired glucose metabolism and the subsequent increase in blood serum glucose concentration. The pathogenic equation for DM presents a complex interrelation of metabolic, genetic and environmental factors, as well as inflammatory mediators. Among the latter, it is mostly unclear whether they reflect the disease process or are simply signs of systemic or local responses to the disease [1].
Currently diabetes is classified into: 1) Type 1 diabetes which is an autoimmune disease influenced by genetic and environmental factor. It is characterized by destruction of pancreatic beta cells mediated by T cells that leads to partial or total absence of insulin secretion. The presence of auto-antibodies associated with diabetes is the most reliable marker of the existence of the disease although there is no evidence that they are the cause of the disease, 2) Type 2 diabetes in which there is a resistance state resulting from inefficient use of insulin by the tissues to regulate blood glucose concentration [2], 3) Gestational diabetes is a different entity and is defined as diabetes diagnosed for first time during pregnancy [3]. Other forms of diabetes have been genetically characterized, for example, onset diabetes of the maturity of the young (MODY) that is a familiar form of diabetes that is inherited in an autosomal dominant mutation associated with beta cells or liver specific genes such glucokinase. Other form well characterized of diabetes is called “secondary diabetes” and include those associated with: pancreatic disease, hemochromatosis, excess steroids or other hormones, certain drugs (e.g., protease inhibitors), genetic defects in insulin action, endocrinopathies, infection, and also forms of diabetes uncommon mediated by immunologic mechanisms. The common symptoms of different forms of diabetes are polyuria, polydipsia and polyphagia with weight loss and in some cases, blurred vision [3].
DM affects about 26 million individuals in America and at least 250 million people worldwide (World Health Organization), causing about 5% of all deaths. Besides, the number of affected people is expected to duplicate by 2030 unless urgent measures are taken [4,5]. Every day, 200 children under 14 years are affected by type 1 diabetes, and this number increases by 3 per cent each year, whereas the analogous increment for preschool children reaches 6 per cent [6]. All these data point out the epidemic character of DM.
2. ANIMAL MODELS FOR DIABETES STUDY
Rats and mice are animals commonly used for studying the effects of diabetes. Type 2 DM can be induced in animal models through dietary modification such as the administration of sucrose, fructose, high fat diet and glucose infusion or through genetic manipulation such as db/db mice, ob/ob mice, Goto-Kakizaki rats, Zucker diabetic rats and BHE rats [7].
On the other hand, type 1 diabetes can be replicated in animal models through genetic modifications i.e. non obese diabetic mice (NOD), which spontaneously develop type 1 diabetes in a manner similar to humans [7]. Other animal models genetically selected are the Bio Breeding rats (BB), in which the pancreatic islets are under the attack of immune T cells, B cells, macrophages and natural killer cells. At approximately 12 weeks of age, these diabetic rats presented weight loss, polyuria, polyphagia, hyperglycemia and insulinopenia. As in humans, if these rats are not treated with exogenous insulin, ketoacidosis is severe and fatal [8]. Another way to obtain experimental animals with type 1 diabetes involves the administration of diabetogenic chemicals such as alloxan or streptozotocin [9,11]. In our laboratory, we have shown that treatment with streptozotocin (STZ) causes alterations in biliary excretion during the first seven days post-injection of the drug, and that biliary function is normalized 10 days after injection [10,12]. This is the reason why studies of liver function during STZ-induced diabetic state should be performed fifteen days after injection of STZ. In our work, STZ-induced diabetes (SID) was achieved by a single dose of STZ (60 mg/kg body weight, i.p., in 50 mM citrate buffer, pH 4.5). Control rats were injected with vehicle alone. Fifteen days after STZ injection, a time when the toxic effect of the drug on the liver has disappeared [9,10], serum glucose levels were tested by means of the glucose oxidase method (Wiener Lab., Rosario, Argentina) in samples of diabetic and control animals. Successful induction of diabetes was defined as a blood glucose level >13.2 mmol/l.
3. DIABETES AND INFLAMMATION
Inflammation represents a protective response that controls infections and promotes tissue repair, but it can also contribute to local tissue damage in a broad spectrum of inflammatory disorders. The inflammatory responses are associated with variations of a broad array of plasma proteins and pro-inflammatory cytokines. The acutephase response is a systemic reaction in which the concentration of a variety of plasma proteins, termed acutephase proteins, may increase or decrease in response to inflammation [13]. Plasma concentrations of acute-phase proteins are largely dependent on the hepatic biosynthesis of such proteins, and changes in their production are influenced by pro-inflammatory cytokines, such as interleukin (IL) IL-1, IL-6 and tumor necrosis factor alpha (TNF-α). These cytokines are produced during the inflammatory process and are principal stimulators of acute-phase proteins and other markers of chronic inflammation commonly detected in cardiovascular diseases, diabetes mellitus, osteoarthritis and rheumatoid arthritis [14,15].
The chronic hyperglycemia can directly promote an inflammatory state, where the increase in cytokines can lead to destruction of the pancreatic beta cells and malfunction of the endocrine pancreas in both type 1 and type 2 diabetes [16]. There is evidence that autocrine insulin exerts protective anti-apoptotic effects on beta cells and inhibits the suppressor of cytokine signaling (SOCS) induced by various cytokines and leads to the apoptosis of the beta cell [17].
Commonly, type 1 and type 2 DM are considered inflammatory processes [18,19] as there is a significant increase in IL-6, IL-18, IL-1 and TNF-α in the blood of patients with this disease [20,21]. Furthermore, chemokines (ligands 2 and 5 chemokines CCL2, CCL5 and CX3CL1), intercellular adhesion molecule-1 (ICAM-1), vascular cell adhesion molecule-1 (VCAM-1) and nuclear transcription factor κB (NFκB) are involved in the development and progression of the disease [22,23].
4. DIABETES AND OXIDATIVE STRESS
Oxidative stress, also referred to as a reactive oxygen species (ROS)/antioxidant imbalance, occurs when the net amount of ROS exceeds the antioxidant capacity. Thus, oxidative stress can occur because of a general increase in ROS generation, a depression of the antioxidant systems, or both. ROS derived from various oxidation pathways can generate products leading to cellular deregulation. ROS mainly includes superoxide anion (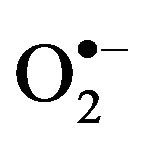 ), hydrogen peroxide (H2O2) and hydroxyl radical (HO.), which results in the production of other free radicals for example, nitric oxide (NO) and peroxinitrite (ONOO.), also called reactive nitrogen species (RNS) [24,25].
), hydrogen peroxide (H2O2) and hydroxyl radical (HO.), which results in the production of other free radicals for example, nitric oxide (NO) and peroxinitrite (ONOO.), also called reactive nitrogen species (RNS) [24,25].  can be generated by specialized enzymes, such as the nicotinamide adenine dinucleotide phosphate (NADPH) oxidase and then can be converted to H2O2 in the presence of superoxide dismutase (SOD). In the presence of high iron (Fe+3), H2O2 can be reduced, resulting in the generation of hydroxyl radical. NO is generated through nitric oxide synthase (NOS).
can be generated by specialized enzymes, such as the nicotinamide adenine dinucleotide phosphate (NADPH) oxidase and then can be converted to H2O2 in the presence of superoxide dismutase (SOD). In the presence of high iron (Fe+3), H2O2 can be reduced, resulting in the generation of hydroxyl radical. NO is generated through nitric oxide synthase (NOS).  can also react with NO to form the highly reactive ONOO· [26].
can also react with NO to form the highly reactive ONOO· [26].
Under normal conditions, the body has a potent antioxidant system to that protects it from the deleterious effects of ROS. When either the production of ROS is increased or the antioxidant defenses inactivated, oxidative stress occurs and ROS can react with cellular macromolecules and enhance the process of lipid peroxidation (LPO), cause DNA damage and/or induce protein and nucleic acid modifications [27,28]. ROS generation has been implicated in the pathogenesis of various disease states, including atherosclerosis and inflammation [29-33].
Chronic hyperglycemia causes increased formation of advanced glycated end products, increased glucose autooxidation and oxidative phosphorylation, which lead to oxidative stress. It is well established that hyperglycemia elicits an increase in ROS production, due to increased input reducing equivalents into the mitochondrial electron transport chain [34]. Thus, hyperglycemia-induced oxidative stress or activation of other pathways may cause tissue damage.
West indicated an increase in oxidative damage in both type 1 and type 2 diabetes as well as deficits in antioxidant defense enzymes and vitamins. It is argued that oxygen, antioxidant defenses, and cellular redox status should be regarded as central players in diabetes [35].
Mitochondrial dysfunction is considered to be a critical component in the development of insulin resistance and type 1 diabetes such as STZ-induced diabetic (SID) rats and mice where hyperglycemia induces an acceleration of hydroxyl radical generation and this has been correlated with the level of thiobarbituric acid thiobarbituric acid reactive compounds (TBARS) reactive compounds as an estimation of LPO [36,37]. In this connection, we have demonstrated that hyperglycemia increases the hydroxyl radical production in the liver of STZ-induced diabetic rats [38]. In addition, the increase in oxidative stress induced by hyperglycemia and inflammation conduces to the development of associated diseases, such as diabetic nephropathy [18,22].
5. NITRIC OXIDE IN TNF-α PATHWAYS AND APOPTOSIS
As stated above, one of the main cytokines released in these inflammatory processes is TNF-α, which can activate signaling pathways associated with cell survival, apoptosis, inflammatory response and cell differentiation. The induction of the responses mediated by TNF-α occurs through the binding of the cytokine to the receptors TNF-R1 and TNF-R2. Both receptors may mediate cell death. TNF-R1 contains a death domain while TNF-R2 does not but it would enhance the cytotoxic effects of TNF-R1. In response to inflammation and infection, TNF-α is produced primarily by immune system cells such as macrophages and lymphocytes [39,40]. The binding of TNF-α to TNF-R1 can promote the activation of NFκB or initiate the activation of caspases, which play a major role in the execution of programmed cell death or apoptosis [41]. NFκB stimulates the expression of genes encoding cytokines (e.g. TNF-α, IL-1, IL-6, IL-2, IL-12, INF-g and CM-CSF), cell adhesion molecules (CAMs), chemokine receptors and inducible enzymes (e.g., COX- 2, iNOS). It also increases the expression of molecules involved in regulating cell proliferation, apoptosis and cell cycle progression, such as the cellular inhibitor of apoptosis protein 1 (c-IAP1), c-IAP2, TNF-receptor-associated factor 1 (TRAF-1), TRAF-2, B-cell lymphocyte/ leukemia-2 (Bcl-2), Fas, c-myc and cyclin D1 [42,43]. It has been found that high levels of glucose can cause apoptosis, in part, through activation of NFκB [44]. It has been shown that high glucose levels activate protein kinase C (PKC) pathway, NFκB and ROS [45-47]. Furthermore, cytokines and bacterial pathogens can activate iNOS and generate large concentrations of NO, through activation of NFκB [7].
6. REACTIVE OXYGEN SPECIES IN MODULATION OF APOPTOSIS
Apoptosis is a tightly regulated form of cell death in which a cell effectively partakes in its own demise. The programmed cell death is an essential physiological process required for normal development and maintenance of tissue homeostasis execution of the death program is characterized by morphological changes which includes blebbing, chromatin condensation, nuclear fragmentation, loss of adhesion and rounding and cell shrinkage. Biochemical features associated with apoptosis include high molecular weight, deoxyribonucleic acid (DNA) fragmentation and/or fragmentation into an oligonucleosomal ladder, phosphatidyl serine externalization, proteolytic cleavage of a number of intracellular substrates and mitochondrial permeability transition pore (MPTP) assembly [48]. Apoptotic cells are detected in organs, such as the liver, during development as well as in healthy adults. Increases of apoptotic cells are present in many chronic inflammatory and autoimmune diseases.
Oxidative stress occurs in cells when the generation of ROS overwhelms the cell’s natural antioxidant defenses. Moreover, the oxidative stress and the redox state of a cell play a pivotal role in regulating apoptosis [49]. The role of ROS in the induction of apoptosis is provided by several studies in which the addition of ROS in low levels induces apoptosis and the observation that various antioxidants can inhibit cell death [50,52]. Oxidative stress can lead to damage of the mitochondrial inner membrane, resulting in MPTP formation and subsequent release of cytochrome c and apoptosis inducing factor from the mitochondria. In the cytosol, cytochrome c complexes with Apaf-1 to activate procaspase 9, which in turn activates downstream effector caspases (3, 6 and 7) [53]. Furthermore, the MPTP is regulated by the relative prevalence of anti-apoptotic (Bcl-2, Bcl-XL) or proapoptotic (Bad, Bax) Bcl-2 family members into the mitochondrial membrane [54].
The initial cellular response to high glucose challenge is the generation of ROS, which rapidly induces apoptotic cell death [55]. It is known that high D-glucose induces endothelial apoptosis through activation of the Bax-caspase proteases pathway. The effectors of apoptosis are now well known to be represented by a family of intracellular cysteine proteases known as caspases [56]. A feature of apoptosis that impinges on caspases is altered mitochondrial function characterized by a reduction in the electrochemical gradient across the mitochondrial membrane and a release of mitochondrial cytochrome c to cytoplasm, and it is inhibited by the presence of Bcl-XL in these organelles [47,57]. Translocation of pro-apoptotic Bax protein into the mitochondrial membrane is accompanied by a significant increase in caspase-3 and caspase-9 activities [58].
7. DIABETES AND LIVER APOPTOSIS
The onset of diabetes is accompanied by development of major biochemical and functional abnormalities in the liver, including alterations in carbohydrate, lipid, and protein metabolism, and changes in antioxidant status [59-62]. The prevalence of hepatobiliary diseases is increased in patients with either type 1 or type 2 diabetes [62,63].
Diabetes is known to be a major disorder in which oxidative stress and TNF-α production have been implicated through several lines of evidence [34,64,66]. ROS and TNF-α have been defined as an autocatalytic mechanism that can lead to programmed cell death (apoptosis) [67].
Regulation of cell death by apoptosis may be another determinant of liver structure and lesion formation [68]. It has become increasingly clear that the process of cell death by apoptosis is a relatively ubiquitous phenomenon in a variety of cell types, including hepatic cells [69].
In a research recently published by us, it is demonstrated that the diabetic state induces an increase of TNF-α and of its receptor TNF-R1 in the liver [65]. Following TNF-α binding to the TNF-R1, an adaptor molecule (TRADD, TNF receptor associated DD protein), is recruited by the dead domain (DD) to form the first protein complex, which also includes TRAF2 [70]. This complex then dissociates from TNF-R1 and forms a different complex in the cytosol, which binds FADD (Fas associated DD protein), and then recruits caspase-8. Cleavage of pro-caspase 8 allows the release of activated caspase-8 [71]. Caspase-8 can cleave Bid to form an active fragment, t-Bid [72]. In the liver of STZ-induced diabetic rats we observed an increase in the pathway that begins with the triggering of receptor TNF-R1 by TNF-α, demonstrated by increased expression and activity of caspase-8 and mitochondrial t-Bid [63]. Pro-apoptotic protein Bid promotes initiation of the mitochondrial death pathway with release of cytochrome c, and activation of effectors caspase-3 that ultimately induce apoptosis [73]. Our results clearly demonstrated that in the liver of STZ-induced diabetic rats induces an enlargement of caspase-3 activity with the consequent increase in the apoptosis index. Besides, the anti–TNF-α (etanercept) treatment produced a declination in the response of receptor TNF-R1 to TNF-α (diminished caspase-8 expression and activity and mitochondrial protein t-Bid, in comparison with SID group) showing a reduction of the cascade of events leading to apoptotic cell death [65].
An early study had demonstrated that the activation of JNK is associated with increased TNF-α induced apoptosis hepatocytes [74]. In this connection, ours studies demonstrated that the diabetes leads to the activation of JNK, inducing an increase of the apoptotic index. Moreover, we have demonstrated that the diminution of TNF-α levels by the treatment with etanercept seems to completely abolish the observed the activation of JNK induced by the diabetic state, leading to the diminution of the apoptosis [63].
The increase of the TNF-α level in the liver of STZinduced diabetic rats leads to a marked up-regulation of the NFκB pathway. NFκB is one of the key transcription factors involved in triggering the cascade of events that allow inflammation and different research groups have demonstrated its activation in the diabetic liver [75,76]. The expression of iNOS is closely related to stimulation of NFκB. Indeed, NFκB recognition sites have been identified in the promoter region of the gene encoding for iNOS.
In the liver of diabetic rats, we found an increase in TNF-α due to an increased expression of iNOS, which led to a high production of NO [65]. Similar results have been reported in different tissues by other authors [77,78]. We observed that the treatment with etanercept, which blocks TNF-α, leads to a decrease in the expression of iNOS, which is increased in the diabetic state. Furthermore, the mentioned treatment reduces the production of NO in the liver of STZ-induced diabetic rats. It has been shown that high concentrations of glucose cause an increase in the expression of iNOS induced by cytokines in rat tissues [79]. Consistent with this, high glucose concentrations do not increase iNOS in the absence of TNF-α [65]. The inhibition of iNOS with a selective inhibitor, such as aminoguanidine, also reduced the production of TNF-α, thus evidencing an interaction between TNF-α pathway and the activity of iNOS. In addition, the treatment with aminoguanidine decreases caspase-8 and caspase-3 activities, resulting in effective blockade of apoptosis in the liver of STZ-diabetic rats [65].
Our studies demonstrate that in the diabetic liver oc-

Figure 1. Proposed scheme for the mechanism involved in the hyperglycemia-induced apoptosis that occurs in type 1 diabetes-induced liver disease. In the diabetic state, the increase in hepatic TNF-α induces activation of NFκB, caspase-8 and JNK, which leads to an increase in apoptotic events. Besides, hyperglycemia increases ROS levels, specifically hydroxyl radical, which initiate mitochondrial apoptosis either directly and/or by acting through Bcl2 family proteins, thus inducing the formation of the mitochondrial permeability transition pore (MPTP), the release of cytochrome c and the activation of caspase 3.
curs apoptosis. Importantly, we have identified that the hydroxyl radical contributes partially to increase mitochondrial Bax protein expression leading to cytochrome c release and caspase-3 activation which are associated with hyperglycemia-induced liver apoptosis. The strong inhibition of the LPO and apoptosis elicited by the treatments with Desferal (potent iron chelator) [73] or Tempol (direct scavenger of hydroxyl radicals) [74] clearly establishes a connection between production of hydroxyl radical, the LPO levels and apoptosis [36].
At the light of previous knowledge and from our own results, we present in Figure 1 a summary of the sequence of events occurring in the hepatocytes of the diabetic liver under hyperglycemic conditions.
8. CONCLUSION
The relevance of the present review is to provide further knowledge about the mechanisms which may contribute to the disease process in the liver during the course of an inflammatory process as it is type 1 diabetes. Regulation of hepatic TNF-α levels and oxidative stress in the diabetic state could be of therapeutic relevance for the improvement or delay of the hepatic complications linked to chronic hyperglycemia.
9. ACKNOWLEDGEMENTS
This work was supported by research grants from CONICET. We especially wish to thank Dr Cecilia Basiglio for language revision.
NOTES