Unusual amplification in polymerase chain reaction for a plasmid containing an insert derived from bovine genomic DNA ()
1. INTRODUCTION
The saliva of various animals contains proline-rich proteins which may play important roles in prevention of mineral precipitation, protection of dietary and digestive proteins from interaction with tannins, and modulation of bacterial colonization on the tooth surface [1-4]. We have isolated a proline-rich protein termed P-B from human saliva and determined its amino acid and nucleotide sequences [5]. Following the finding of Strawich and Glimcher that a homologous protein is present in bovine tooth germ [6], we started a study on bovine P-B [7]. In the course of cloning bovine cDNA for P-B, we found a segment of E. coli genomic DNA in bovine tooth germ mRNA encoding P-B [8]. Consequently, we examined the possible occurrence of E. coli DNA-derived nucleotide sequences in bovine genomic DNA using a plasmid library constructed from EcoRV digests of the bovine DNA. Results so far have failed to indicate the presence of E. coli DNA in the bovine nucleotides examined, but they revealed unusual amplification by the polymerase chain reaction (PCR). We report here findings to suggest that a specific nucleotide sequence in combination with the palindrome structure can promote preferential hybridization between a short nucleotide and its complementary structure resulting in the generation of unexpected PCR products.
2. MATERIALS AND METHODS
2.1. Materials
The LA PCR kit, agarose (H14 TAKARA), restriction enzymes, and SYBR Green I Nucleic Acid Gel Stain were purchased from Takara Bio Inc. (Otsu, Japan); SynerGel for use with agarose, from Diversified Biotech Inc. (Boston, MA, USA); Bovine genomic DNA and Perfectly Blunt Cloning Kits with the plasmid pT7Blue vector, from Novagen, Inc. (Madison, WI, USA); the GenElute Five-minutes Plasmid Miniprep Kit and DNA standard for gel electrophoresis, from Sigma-Aldrich Japan, Inc. (Tokyo, Japan); and the Big Dye Terminator cycle sequencing kit, Applied Biosystems Japan Ltd. (Tokyo, Japan).
Primers used were as follows:
M13-47: 5’-CGCCAGGGTTTTCCCAGTCACGAC- 3’ RV-M: 5’-GAGCGGATAACAATTTCACACAGG-3’ T7: 5’-TAATACGACTCACTATAGGG-3’ pT7Blue9: 5’- GATTACGCCAAGCTCTAATA-3’ HindR: 5’-AAGCTTGCATGCCTGCAGGT-3’ B6C2985: 5’-ACCCGGGGATCCGATATCTT-3’
M13-47, RV-M, and T7 Bos BestTM sequencing primers were obtained from Takara Bio. Inc. The others were custom-made products of Invitrogen Life Technologies (Tokyo, Japan). Their positions in the plasmid B6 are indicated in Figure 1 and/or Figure 6(b).
2.2. Agarose Gel Electrophoresis
Agarose gel (1%) was used for the electrophoresis of nucleotide sequences longer than 1000 bp. For shorter sequences, gels prepared from 0.7% agarose and 1.2% Syner gel according to the manufacturer’s instructions were used. Nucleotides were visualized by staining with SYBR Green I.
2.3. Plasmid Library
Bovine genomic DNA (50 μg) was digested with EcoRV (450 U) in 250 μl of universal buffer H (Takara) and fractionated by agarose gel electrophoresis. Using a gel section containing 3000 - 4200 bp and the vector pT7Blue, we constructed a clone-pool library in 96-well plates. From the agar culture plate containing E. coli with plasmids from each well, 10 colonies each were removed from wells A1-H12 to prepare plasmid mixtures termed a1-h12, respectively.
2.4. PCR
PCR was performed using a long and accurate (LA) PCR kit suitable for amplification of long DNA according to the manufacturer’s instructions. Plasmid mixtures, cloned plasmids or restriction enzyme digests were subjected to PCR amplification (40 cycles of denaturation at 95˚C for 30 sec, annealing at 55˚C for 30 sec, and elongation at 72˚C for 1 min) in a total volume of 10 μl (1 μl each of forward and reverse primers (2 μM), 10 ng of template, 5 μl of One Shot LAPCR Mix, and water to give a final volume of 10 μl) using MyCycler Thermal Cycler (BIO-RAD, Hercules, CA, USA).
2.5. Restriction Enzyme Digestion
Plasmids were digested with HindIII or EcoRI at 37˚C overnight according to the manufacturer’s instructions.
2.6. Cloning of B6
B6 was cloned from well G12 according to methods described previously [5,8].
2.7. Nucleotide Sequence
Nucleotide sequences were analyzed by the dye terminator method on an ABI PRISM 310NT Genetic Analyzer (Applied Biosystems Japan Ltd., Tokyo, Japan) as described previously [5,8].
3. RESULTS
In this study, we prepared 96 plasmid mixtures (a1- h12) expected to contain 3000 - 4200 bp inserts derived from bovine genomic DNA from 10 colonies of E. coli with the vector pT7Blue. The plasmid mixture derived from well G12 was named g12. According to the assumed plasmid structure shown in Figure 1, we expected PCR using the primers M13-47 and RV-M to give a nucleotide product larger than 3000 bp. However, the major product was much smaller (ca. 300 bp) as shown in

Figure 1. A schematic overview of the assumed structure of a constructed plasmid, g12. A plasmid library was prepared from EcoRV digests (ca. 3000 bp) of bovine genomic DNA ligated with the vector pT7Blue. The plasmid mixture g12 was selected from 10 colonies of E. coli. Restriction enzyme sites are shown. The positions of the sequence used for designing primers are indicated by arrows.
Figure 2(a). If the RV-M or M13-47 sequence is present in the insert near the EcoRV site, PCR using RV-M and M13-47 might give a product of such size, but PCR with the HindIII digest of g12 would not (see Figure 1). When PCR was carried out using the HindIII digest of g12, the product was ca. 150 bp (Figure 2(b)), suggesting it to be unlikely that the RV-M or M13-47 sequence is present in the insert.
The nucleotide sequence of ca. 150 bp termed pn-150 thus obtained was determined as shown in Figure 3(a). This finding indicated that pn-150 contained the 19-base sequence TTAAAGCAGTAGCGTATTG, between the EcoRV site extending from the M13-47 sequence in the vector and the vector sequence extending from RV-M, and that this portion was not derived from pT7Blue (Figure 3(a)). Therefore, this section was considered to be derived from bovine genomic DNA.
To explain this unexpected result, we cloned the plasmid which gave a 300-bp PCR product using g12, and obtained a plasmid termed B6. When we digested B6 with HindIII on the basis that pT7Blue contains a single HindIII site, we obtained a band of ca. 6000 bp (Figure 4(a)), indicating that B6 was of the expected size. PCR using the intact B6 and a combination of forward (M13- 47) and reverse (RV-M) primers gave a major product of ca. 300 bp termed b6-300 (Figure 4(b)). Thus, the plasmid B6 gave a 300-bp PCR product in the plasmid mixture g12. B6 gave products of less than 300 bp when we used primers to bind inner positions directed at the insert instead of the combination of M13-47 and RV-M (Figure 5, see also Figures 1 and 6(b)), indicating that amplifycation similar to that with primers of M13-47 and RV-M occurred using other primers under the present conditions.
A portion of bovine genomic DNA in B6 was sequenced and registered as “Bos taurus DNA, palindrome sequence region” in NCBI GenBank (AB511281) (see Figure 6(a)). The nucleotide sequence of B6 around the EcoRV site together with 124 bp from RV-M and 71 bp from the M13-47 side is shown in Figure 6(b). The sites for which primers were designed are also indicated (see also Figure 1).
Based on the structure of B6, we explain how the HindIII digest of g12 gave the PCR product shown in Figure 3(a) (Figure 3(b)). When the nucleotide chain elongated from RV-M reached C–87-A-T–85 (numbering based on the sequence shown in Figure 6(b)), this portion hybridized with G2946(-) -T- A2948(-) of the chain which had been elongated from the 5’-end of M13-47, and vice versa (Figure 3(b)). Subsequent elongation of each chain proceeded by reading the sequence of the counter chain as shown in Figure 3(b) to give pn-150.
When the primer M13-47 or RV-M was used to determine the nucleotide sequence of b6-300 (see Figure 4(b)), we were unsuccessful for unknown reasons. When T7 was used for sequencing, a partial sequence of b6-300
 (a) (b)
(a) (b)
Figure 2. (a) Gel electrophoresis of the products of PCR using g12 as a template and the primers M13-47 and RV-M. Products of PCR (lane 1) and a size marker with multiples of 100 bp (lane 2); (b) Nucleotide (pn-150) produced by PCR of the HindIII digest of g12 (lane 1) and the size marker (lane 2).
 (a)
(a) (b)
(b)
Figure 3. (a) Nucleotide sequence of pn-150. Arrows indicate primer sequences. The bold-face nucleotides show the structure presumably derived from bovine genomic DNA. The EcoRV site is boxed. Base numbers are based on those of the structure shown in Figure 6(b); (b) A possible mechanism for PCR amplification of pn-150. After the M13-47 primer (M) bound to the complintary sequence starting at G3044 in the template, elongation continued to make A1-chain with a terminal -A-T-G2946(-) structure. B1-chain was produced by initial binding of RV-M (R) to the template structure starting at G–124, followed by elongation ill the completion of the trinucleotide structure of -C-A-T–85(-) (Step 1). After hybridization between G2946(-)-T-A and C-A-T–85 (Step 2), A1-chain was elongated according to the sequence of B1 chain to complete A-chain and vice versa (Step 3), resulting in the formation of pn-150. The pink-colored segment represents the seence contained in B6 and green-colored sequences are those associated with the vector.
 (a) (b)
(a) (b)
Figure 4. (a) Agarose-gel electrophoresis of the HindIII digest of B6 (lane 1) and a size marker (lane 2); (b) Agarose-gel electrooresis of the nucleotide (b6-300) produced by PCR using the HindIII digest of B6 and primers M13-47 and RV-M (lane 1) and a size marker (lane 2).
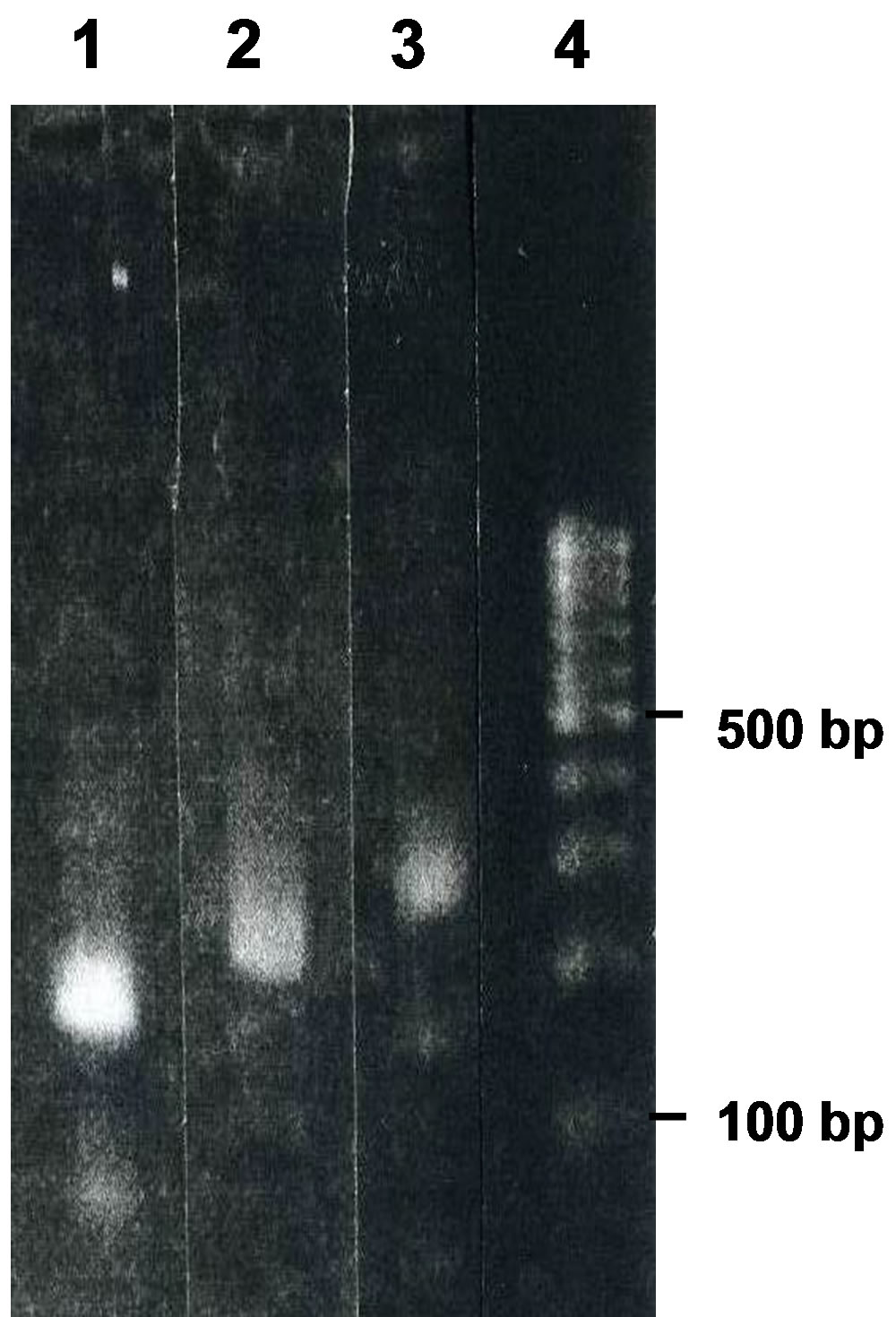
Figure 5. Agarose-gel electrophoresis of the nucleotides produced by PCR using B6 and the primers M13-47 and HindR (lane 1), B6 and the primers M13-47 and pT7Blue9 (lane 2), B6 and the primers M13-47 and RV-M (lane 3), and a size marker (lane 4).
 (a)
(a) (b)
(b)
Figure 6. (a) Single chain of the nucleotide sequence of the cloned plasmid B6 containing sequences derived from bovine genomic DNA and the vector pT7Blue. EcoRV sites are indicated in bold face. The sequence of G1-T870 is the same as the complementary structure of C2973-A2104; (b) Nucleotide sequence of B6 around the EcoRV site (boxed) used as the cloning site. Numbering was started at G1 of the EcoRV site. The 5’-terminal was assigned as −124 and the 3’-terminal as 3044. Numbering for the counterchain was presented by addition of a mark (-) after a number as exemplified by 3044(-) for its 5’-terminal. The sequences used as primers are indicated by arrows. The HindIII site is indicated in bold face. The blue-colored sequence represents B6.
was determined as shown in Figure 7(a). The finding indicated that after a portion of T61-C-A-C64 in the growing chain hybridized to A37-G-T-G34 in the growing counter chain, the extension continued in the 3’direction by reading the sequence of the counter chain preformed by PCR (Figure 7(b)). The finding suggests that two newly synthesized chains which had started from the primer T7 were hybridized to give a double strand polynucleotide shown in Figure 7(a), which represents an inner portion of the polynucleotide of b6-300.
When PCR was performed using the EcoRI digest of B6 as the template and primers B6C2985 and M13-47, a product of ca. 110 nucleotides was amplified and termed b6-110 (Figure 8(a)). The nucleotide b6-110 was sequenced as shown in Figure 8(b). It is suggested that preferential hybridization occurred between the 5-base
 (a)
(a)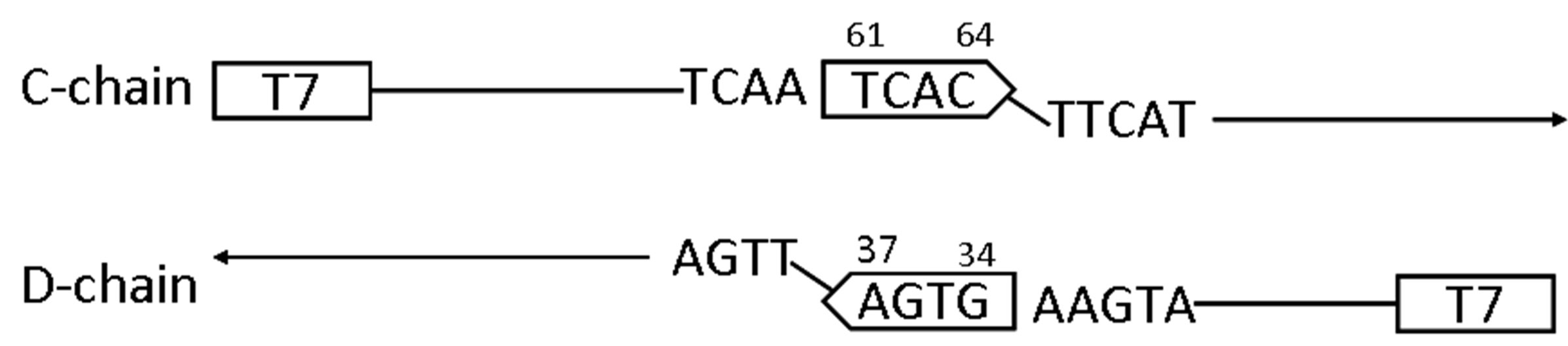 (b)
(b)
Figure 7. (a) The partial nucleotide sequence of b6-300 as determined using the primer T7. The sequence used as the primer is indicated by arrows. The 4-bp structures presumably involved in the initial hybridization are boxed. The EcoRV sites are shown in bold face; (b) A possible mechanism for generation of the portion contained in b6-300. C-chain was elongated from T7 to form the tetranucleotide -T61-C-A-C64 by reading the template and D-chain was elongated from the same primer to form A37-G-T-G34-. After initial hybridization between these tetranucleotides, subsequent elongation was started by reading the sequence of the respective counterchain to complete a double strand structure between Cand D-chains.
nucleotide C2926(-)-T-G-G-C2922(-) and the complementary G3002(-)-A-C-C-G3006(-) as indicated in Figure 8(c), and subsequent elongation was achieved by reading the sequence of the counter chain.
4. DISCUSSION
PCR is useful for amplifying nucleotides having a sequence expected from the use of forward and reverse primers [9]. However, as shown in Figures 3, 7, and 8, PCR gave unexpected results under certain conditions. In the present case, when the two chains were elongated by reading the sequence of the respective template until the completion of a certain hybridizable short (3 - 5) nucleotide structure, subsequent elongation was started by reading the sequence of the counter nucleotide chain that had been elongated from the respective template. The short nucleotide structure participating in the initial hybridization was present at multiple positions of the template used. It is possible that elongation of the chain by reading the template was interrupted once before the completion of amplification due to the template’s palindrome region which had formed a double strand structure during PCR as exemplified by a model for the process during production of b6-110 (Figure 8(d)). Then, the accumulated short nucleotides were hybridized each other through the complementary nucleotide sequence to start elongation by reading the sequence of the counter chain
preformed during PCR.
Such an unusual amplification generated under certain conditions in a DNA sequence may be one of the mechanisms for the genetic recombination found in our previous study [8]. Study to examine possible occurrence of E. coli DNA-derived nucleotide sequences in bovine genomic DNA is now in progress.