Multivariate Statistical Analysis of Large Datasets: Single Particle Electron Microscopy ()
Received 5 December 2015; accepted 28 August 2016; published 31 August 2016

1. Introduction
The electron microscope (EM) instrument, initially developed by Ernst Ruska in the early nineteen thirties [1] , became a routine scientific instrument during the nineteen fifties and sixties. With the gradual development of appropriate specimen-preparation techniques, it proved an invaluable tool for visualizing biological structures. For example, ribosomes, originally named “Palade particles”, were first discovered in the nineteen fifties in electron microscopical images [2] . In the nineteen sixties and seventies, the early days of single-particle electron microscopy, the main specimen preparation approach used for investigating the structure of biological macromolecules was the negative stain technique in which the samples were contrasted with heavy metal salts like uranyl acetate [3] [4] . In those days, the standard way of interpreting the structures was to come up with an intuitively acceptable three-dimensional arrangement of subunits that would fit with the observed (noisy) molecular images.
Around 1970, in a number of ground-breaking publications, the idea was introduced by the group of Aaron Klug in Cambridge of using images of highly symmetric protein assemblies such as helical assemblies or icosahedral viral capsids [5] [6] to actually calculate the three-dimensional (3D) structures of these assemblies. The images of these highly symmetric assemblies can often be averaged in three dimensions without extensive pre- processing of the associated original images. Averaging the many unit cells of a two-dimensional crystal, in combination with tilting of the sample holder gave the very first 3D structure of a membrane protein [7] . Electron tomography of single particles had been proposed by Hoppe and his co-workers [8] , however, due to the radiation-sensitivity of biological macromolecules to electrons, it is not feasible to expose a biological molecule to the dose required to reveal the 3D structure from one hundred different projection images of the same molecule.
For other types of complexes, no methods were available for investigating their 3D structures. The vast majority of the publications of those days thus were based on the visual recognition of specific molecular views and their interpretation in terms of the three-dimensional of the macromolecules. For example, a large literature body existed on the 3D structure of the ribosome-based antibody labelling experiments [9] [10] . The problem with the visual interpretation of two-dimensional molecular images was obviously the lack of objectivity and reproducibility in the analyses. Based on essentially the same data, entirely different 3D models for the ribosome had been proposed [9] . There clearly was a need for more objective methods for dealing with two-dimensional images of three-dimensional biological macromolecules.
Against this background, one of us (MvH), then at the university of Groningen, and Joachim Frank, in Albany NY, started a joint project in 1979 to allow for objectively recognizing specific molecular views in electron micrographs of a negative stain preparation (see Appendix). This would allow one to average similar images prior to further processing and interpretation. Averaging is necessary in single-particle processing in order to improve the very poor signal-to-noise ratios (SNR) of direct, raw electron images. Averaging similar images from a mixed population of images, however, only makes sense if that averaging is based on a coherent strategy for deciding which images are sufficiently similar. We need good similarity measures between images such as correlation values or distance criteria for the purpose. Upon a suggestion of Jean-Pierre Bretaudière (see Appendix), the first application of multivariate statistical analysis (MSA) emerged in the form of correspondence analysis [11] [12] . Correspondence Analysis (CA) is based on chi-square distances [13] - [15] , which are excellent for positive (histogram) data. In retrospect, other distances are more appropriate for use in transmission electron microscopy (see below) but let us not jump ahead.
A most fundamental specimen-preparation development strongly influenced the field of single-particle electron microscopy: In the early nineteen eighties the group around Jacques Dubochet at the EMBL in Heidelberg brought the “vitreous-ice” embedding technique for biological macromolecules to maturity [20] . Already some 25 years earlier, the Venezuelan Humberto Fernández-Morán suggested the idea of rapid freezing biological samples in liquid helium in order to keep them hydrated [21] . Marc Adrian and Jacques Dubochet brought the rapid-freezing technique to maturity by freezing samples in liquid ethane and propane, cooled au-bain-marie in liquid nitrogen [20] . This is the practical approach that is now used routinely (for a review see [22] ). The vitreous-ice specimen preparation technique represented a major breakthrough in structural preservation of biological specimens in the harsh vacuum of the electron microscope. With the classical dry negative-stain preparation technique-the earlier standard-the molecules tended to distort and lie in just a few preferred orientations on the support film. The vitreous-ice, or better, “vitreous-water” technique greatly improved the applicability of quantitative single particle approaches in electron microscopy.
The instrumental developments in cryo-EM over the last decades have been substantial. The new generation of electron microscopes-optimized for cryo-EM-are computer controlled and equipped with advanced digital cameras (see, for example: [23] ). These developments enormously facilitated the automatic collection of large cryo-EM data sets (see, for example [24] ). A dramatic improvement of the instrumentation was the introduction of direct electron detectors, which represent an efficiency increase of about a factor four with respect to the previous generation of digital cameras for electron microscopy [25] [26] . This development also allowed for data collection in “movie mode”: Rather than just collecting a single image of a sample, one collects many frames (say 5 to 50 frames) which can later be aligned and averaged to a single image [27] [28] . The sample is moving continuously over many Ångstrom during the electron exposure which has the effect of a low-pass filter on the single averaged image; “frame alignment” can largely compensates for this effect. Single particle cryo-EM thus recently went through a true “Resolution revolution” [29] .
These newest developments facilitate the automatic collection of large dataset such as are required to bring subtle structural/functional differences existing within the sample to statistical significance. The introduction of movie-mode data collection by itself already increased the size of the acquired data set by an order of magnitude. Cryo-EM datasets already often exceed 5000 movies of each 10 frames, or a total of 50,000 frames of each 4096 × 4096 pixels. This corresponds to 1.7 Tb when acquired at 16 bits/pixels (or 3.4 Tb for 32 bit/pixel). From this data one would be able to extract ~1,000,000 molecular images of each, say, 300 × 300 pixels. These 1,000,000 molecular images represent 100,000 molecular movies, of each 10 frames. The MSA approaches are excellent tools to reveal the information hidden in our huge, multi-Terabyte datasets.
2. The MSA Problem: Hyperspaces and Data Clouds
First, One image is simply a “measurement” that can be seen as a collection of numbers, each number repre- senting the density in one pixel of the image. For example, let us assume the images we are interested in are of the size 300 × 300 pixels. This image thus consists of 300 × 300 = 90,000 density values, starting at the top left with the density of pixel (1, 1) and ending at the lower right with density of pixel (300, 300). The fact that an image is intrinsically two-dimensional is not relevant for what follows. What is relevant is that we are trying to make sense out of a large number of comparable measurements, say 200,000 images, all of the same size, with pixels arranged in the same order. Each of these measurements can be represented formally by a vector of numbers F(a), where a is an index running over all pixels that the image contains (90,000 in our case). A vector of numbers can be seen as a line from the origin, ending at one specific point of a multi-dimensional space known as hyperspace.
The entire raw data set, in our example, consists of 200,000 images of each 90,000 pixels or a total of 18 × 109 pixel density measurements. In the hyperspace representation, this translates to a cloud of 200,000 points in a 90,000 dimensional hyperspace, again consisting of 18 × 109 co-ordinates. Each co-ordinate corresponds to one of the original pixel densities, thus the hyperspace representation does not change the data in any way. This type of representation is illustrated in Figure 1 for a data set simplified to the extreme, where each image consists of just two pixels.
The basic idea of the MSA approach is to optimize the orthogonal co-ordinate system of the hyperspace to best fit the shape of the cloud. We wish to rotate (and possibly shift) the co-ordinate system, such that the first axis of the rotated co-ordinate system will correspond to the direction of the largest elongation of the data cloud. In the simplistic (two-pixel images) example of Figure 1, the largest elongation of the cloud is associated with the average density in the two pixels increasing together. That main direction points from the lower left of the illustration (both pixels have low density) to the top right (both pixels high density). The remaining direction is perpendicular to the first one (also indicated in the illustration) but that direction describes only small modulations with respect to the main trend of the data set and may be ignored. The power of the MSA approach lies in this data reduction. It allows us to then concentrate on the most important trends and variations found in a complex data set and ignore all the other sources of fluctuations (which in EM usually is just noise). We thus enormously reduce the total amount of data into just a few main “principal” components, which show the most significant variations of the data cloud.
![]()
Figure 1. Hyperspace representation of an (extremely) simple set of images, each image consisting of only two pixels. Thus, two numbers completely determine each raw image in this minimalistic data set. Each image is fully represented by a single point in a two-dimensional hyperspace. Together, these points form a data “cloud”. The cloud has a shape as indicated in this example. The main purpose of “MSA” approaches is to optimally adapt the co-ordinate system of this hyperspace to the shape of the cloud, as indicated by the blue arrows in this picture. The shape of the data cloud indicates that the largest source of variations in this data set is that of the densities of both pixels increasing together. That single (rotated) direction describes most of the differences between the images of the dataset.
Concentrating on the main direction of variations within the data, in the example of Figure 1, reduces the problem from a two-dimensional one to just a one-dimensional problem. This reduction of dimensionality can take dramatic proportions with real EM data sets. In the case of a data set of two hundred thousand images of 300 × 300 pixels, typically some 50 - 200 orthogonal directions suffice to describe the most significant (largest) sources of variations within the data. Each image is then no longer described by the 90,000 density values of its pixels, but rather by just its 50 - 200 co-ordinates with respect to those main directions of variations. This represents a reduction in the dimensionality of the data by more than three orders of magnitude. After this data reduction, it becomes feasible to perform exhaustive comparisons between all images in the data set at a reasonable cost. The main orthogonal directions of variations within the data are known as “eigenvectors”; the variance that each eigenvector describes is known as the corresponding eigenvalue. Because each eigenvector corresponds to a point in hyperspace these, in our case, have the character of “image” and are therefore usually referred to as “eigenimages”.
3. Distances and Correlations: The Choice of the Metric
Multivariate Statistical Analysis is all about comparing large sets of measurements and the first question to be resolved is how to compare them. What measure of similarity one would want to use? The concepts of distances and correlations between measurements are closely related, as we will see below. Different distance and associated correlation criteria are possible depending on the metric one chooses to work with. We will start with the simplest and most widely used metric: The classical Euclidean metric.
3.1. Euclidean Metrics
The classical measure of similarity between two measurements F(a) and G(a) is the correlation or inner product (also known as the covariance) between the two measurement vectors:
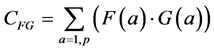 (1)
(1)
The summation in this correlation between the two vectors is over all possible values of a, in our case, the p pixels of each of the two images being compared. (This summation will be implicit in all further formulas; implicit may also be the normalization by 1/p). Note that when F and G are the same (F = G), this formula yields the variance of the measurement: the average of the squares of the measurement:
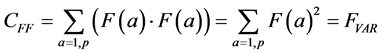 (1a)
(1a)
Closely related to the correlation is the Euclidean squared distance between the two measurements F and G:
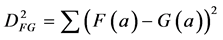 (2)
(2)
The relation between the correlation and the Euclidean distance between F and G becomes clear when we work out Equation (2):
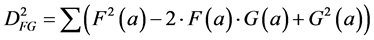 (3)
(3)
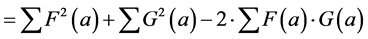
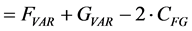 (4)
(4)
In other words, the Euclidean square distance between the two measurements F and G, is a constant (the sum of the total variances in F and in G, FVAR + GVAR, respectively, minus twice CFG, the correlation between F and G. Thus correlations and Euclidean distances are directly related in a simple way: the shorter the distance between the two, the higher the correlation between F and G; when their distance is zero, their correlation is at its maximum. This metric is the most used metric in the context of multivariate statistics; it is namely the metric associated with Principal Components Analysis (PCA, see below). Although this is a good metric for signal processing in general, there are some disadvantages associated the with use of pure Euclidean metrics.
One disadvantage of Euclidean distances and correlations are their sensitivity to a multiplication by a constant. For example, suppose the two measurements F and G have approximately the same variance and one would then multiply one of the measurement, say F(a), with the constant value of “10”. A multiplication of a measurement by such a constant does not change the information content of that measurement. However, the correlation value CFG between the measurements F and G (Equation (1)) will increase by a factor 10. The Euclidean square distance will, after this multiplication, be totally dominated by the FVAR term which will then be one hundred times larger than the corresponding GVAR term in (Equation (4)).
A further problem with the Euclidean Metric (and with all other metrics discussed here) is the distorting influence that additive constants can have. Add a large constant to the measurement F and G, and their correlation (Equation (1)) and Euclidean distance (Equations (2)-(4)) will be fully dominated by these constants, leaving just very small modulations associated with the real information content of each of F and G. A standard solution to these problems in statistics is to correlate the measurements only after subtracting the average and normalising them by the standard deviation of each measurement. The correlation between F and G thus becomes:
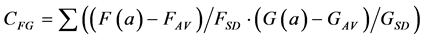 (5)
(5)
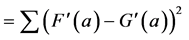 (5’)
(5’)
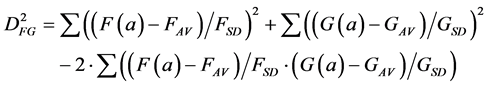 (6)
(6)
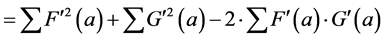 (6’)
(6’)
This normalisation of the data is equivalent to replacing the raw measurements F(a) and G(a) by their normalised versions 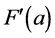 and
and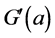 :
:
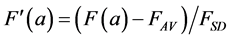 (7)
(7)
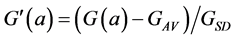 (8)
(8)
These substitutions thus render the Euclidean metrics correlations and distances (Equations (5) and (6)) to exactly the same form as the original ones (Equations (1) and (2)).
3.2. Pretreatment of the Data
Interestingly, it is a standard procedure to “pretreat” EM images prior to any processing and during the various stages of the data processing and this routine is, in fact, a generalisation of this standard normalisation in statistics discussed above. During the normalisation of the molecular images [30] one first high-pass filters the raw images to remove the very low spatial frequencies. These low spatial frequencies are associated mainly with long-range fluctuations in the image density on a scale of ~20 nm and above. Such long-range fluctuations are not directly related to the structural details we are interested in, and they often interfere with the alignment procedures traditionally required to bring the images in register.
The high-pass filtering is often combined with a low pass filter to remove some noise in the high-frequency range, again, trying to reduce structure-unrelated noise. These high spatial frequencies, however, although very noisy, also contain the finest details one hopes to retrieve from the data. For a first 3D structure determination, it may be appropriate to suppress the high frequencies. During the 3D structural refinements, the original high frequency information in the data may later be reintroduced. The overall filtering operation is known as “band- pass” filtering.
The high-pass filtering indeed removes the very-low frequencies in the data but that also has the effect of setting the average density in the images to a zero value. In cryo-EM the predominant contribution to the image generated in the transmission electron microscope (“TEM”) is phase contrast. In phase contrast, the very low frequency information in the images is fundamentally not transmitted by the phase-contrast imaging device, because that area of the back focal plane of the imaging device is associated with the image of the illuminating source or “zero order beam” [31] [32] . Therefore, in cryo-EM the image information is modulated around the value of zero. The average phase contrast modulations in an electron micrograph, over a sufficiently large area, are thus necessarily always essentially zero. Strictly speaking the background value in a phase-contrast image is not “zero” but rather the average density in the image, a value which is not normally experimentally available. The band-pass filtering allows one to concentrate on a range of structural details sizes that are important at the current level of processing or analysis. It has a clear relation to the standard normalization of measurements, as in Equations (7) and (8), but has a much richer range of applications.
3.3. Chi-Square Metrics (χ2-Metrics)
As was mentioned above, the first applications of MSA techniques in electron microscopy [11] [12] focused on “correspondence analysis” (CA) [13] - [15] which is based on Chi-square metrics (c2-metric). Chi-square distances are good for the analysis of histogram data, that is per definition positive. The c2 correlation and distance are, respectively:
![]() (9)
(9)
and,
![]() (10)
(10)
With the c2-metrics, the measurements F and G are normalized by the average of the measurements:
![]() (11)
(11)
![]() (12)
(12)
Substituting the normalized measurements (11) and (12) into formulas (9) and (10) brings us again back to the standard forms (1) and (2) for the correlation and distance.
Why this normalisation by the average? Suppose that of the 15 million inhabitants of Beijing, 9 million own a bicycle, and 6 million do not own a bicycle. Suppose also that we would like to compare these numbers with the numbers of cyclists and non-cyclists in Cambridge, a small university city with only 150,000 inhabitants. If the corresponding numbers for Cambridge are 90,000 bicycle owners versus 60,000 non-owners, then the c2 distance (10) between these two measurements is zero, in spite of the 100 fold difference in population size between the two cities. This metric c2 is thus well suited for studying histogram-type of information.
Interestingly, with the c2-distance, the idea of subtracting the average from the measurements in already “built in”, and leads to identically the same distance (10) as can be easily verified:
![]() (10a)
(10a)
This illustrates that the c2-metrics are oriented towards an analysis of positive histogram data, that is, towards data were the role of the standard deviation FSD and of the average FAV in Equation (7), are both covered by the average of the measurement FAV. Although this leads to nice properties in the representation of the data (see below) problems arise when the measurements F(a) are not histogram data but rather a non-positive signal. The normalisation by the average FAV rather than by the standard deviation FSD may then lead to an explosive behaviour of ![]() when the average of the measurements gets close to zero.
when the average of the measurements gets close to zero.
3.4. Modulation Metrics
As was discussed above, a disadvantage of the Euclidean distance and of the associated correlation is the sensitivity to multiplication of one of the data vectors by a constant. The c2-metric does not have this sensitivity to a multiplicative factor through its normalisation of the measurements by their average. In contrast to histogram data, however, in signal and image processing the measurements need not be positive. Signals often have (or are normalized to) zero average density as discussed in the paragraph above. The c2-metric, when applied to signal-processing measurements is associated with a fundamental problem. This problem in the application of correspondence analysis per se to electron microscopical data was realised soon after its introduction in electron microscopy, and a new “modulation”-oriented metric was introduced to circumvent the problem [33] . With the modulation distance, one divides the measurements by their standard deviation (“SD”). The correlation value and the square distance with modulation metrics thus becomes:
![]() (13)
(13)
and,
![]() (14)
(14)
With
![]() (15)
(15)
and,
![]() (16)
(16)
The MSA variant with modulation distances we call “modulation analysis”, and this MSA technique shares the generally favourable properties of CA, yet, as is the case in PCA, allows for the processing of zero-aver- age-density signals.
4. Matrix Formulation: Some Basics
We have thus far considered an image (or rather any measurement) as a vector named F(a) or as a vector named G(a). We want to study large data sets of n different images (say: 200,000 images), each containing p pixels (say 90,000 pixels). For the description of such data sets we use the much more compact matrix notation. For completeness, we will repeat some basic matrix formulation allowing the first time reader to remain within the notation used here.
4.1. The Data Matrix
In matrix notation we describe the whole data set by a single symbol, say “X”. X stands for a rectangular array of values containing all n × p density values of the data set (say: 200,000 × 90,000 measured densities). The matrix X thus contains n rows, one for each measured image, and each row contains the p pixel densities of that image:
![]() (17)
(17)
4.2. Correlations between Matrices
This notation is much more compact than the one used above because we can, for example, multiply the matrix X with a vector G (say an image with p pixels) to yield a correlation vector C as in:
![]() (18)
(18)
The result of this multiplication ![]() is a vector C, which has n elements, and any one element of C, say Ci has the form:
is a vector C, which has n elements, and any one element of C, say Ci has the form:
![]() (19)
(19)
This sum is identical to the correlation or inner product calculation presented above for the case of the Euclidean metrics (Equation (1)). Explained in words: one here multiplies each element from row i of the data matrix X with the corresponding element of vector G to yield a vector C, the n elements of which are the correlations (or inner products or “projections”) of all the images in the data set X with the vector G.
An important concept in this matrix formulation is that of the “transposed” data matrix X denoted as XT:
![]() (20)
(20)
In XT, the transposed of X, the columns have become the images, and the row have become what first were the columns in X. Similar to the multiplication of the matrix X with the vector G, discussed above, we can calculate the product between matrices, provided their dimensions match. We can multiply the X with XT because the rows of X have the same length p as the columns of XT:
![]() (21a)
(21a)
This matrix multiplication is like the earlier one (18) of the (n × p) matrix X with a single vector G (of length p) yielding a vector C of length n. Since XT is itself a (p × n) matrix the inner-product operation is here applied to each column of XT separately, and the result thus is an (n × n) matrix An. Note that each element of the matrix A is the inner product or co-variance between two images (measurements) of the data set X. The n diagonal elements of An contain the variance of each of the measurements. (The variance of a measurement is the co-vari- ance of an image with itself). The sum of these n diagonal elements is the total variance of the data set, that is, the sum of the variances of all images together, and it is known as the trace of A. The matrix A is famous in multivariate statistics and is called the “variance co-variance matrix”. Note that we also have in the conjugate representation (see below):
![]() (21b)
(21b)
4.3. Transposing a Product of Matrixes
Note that the matrix An (Equation (21a)) is square and that its elements are symmetric around the diagonal: therefore its transposed is identical to itself (![]() ). The transposed of the product of two matrices is equal to the product of the transposed matrices but in reverse order as in:
). The transposed of the product of two matrices is equal to the product of the transposed matrices but in reverse order as in:![]() . We thus also have:
. We thus also have:
![]() (21c)
(21c)
4.4. Rotation of the Co-Ordinate System
In the introduction of the MSA concepts above, we discussed that we aim at rotating the Cartesian co-ordinate system such that the new, rotated co-ordinate system optimally follows the directions of the largest elongation of the data cloud. Like the original orthogonal co-ordinate system of the image space, the rotated one can be seen as a collection of, say q orthogonal unit vectors (vector with normalized length of 1), and the columns of matrix U:
![]() (22)
(22)
To find the co-ordinates of the images with respect to the new rotated co-ordinate system we simply need to project the p-dimensional image vectors, stored as the rows in the data matrix X, onto the unit vectors stored as the columns of the matrix U:
![]() (23)
(23)
Each row of the n × q matrix Cimg again represents one full input image but now in the rotated co-ordinate system U. We happen to have chosen our example of n = 200,000 and p = 90,000 such that n > p. That means that for the rotated co-ordinate system U we can have a maximum of p columns spanning the p-dimensional space (in other words: q ≤ p). The size of Cimg will thus be restricted to a maximum size of n × q. We may choose to use values of q smaller than p, but when q = p, the (rotated) co-ordinate matrix Cimg will contain all the information contained in the original data matrix X.
4.5. An Orthonormal Co-Ordinate System
In an orthonormal co-ordinate system the inner products between the unit vectors spanning the space is always zero, apart from the inner product between a unit vector and itself, which is normalised to the value 1. This orthonormality can be expressed in matrix notation as
![]() (24)
(24)
We have already seen the matrix U above (equation (22)); the matrix UT is simply the transposed of U. The matrix Iq is a diagonal matrix, meaning that it only contains non-zero element along the diagonal from the top-left to the lower right of this square matrix. All these diagonal elements have a unity value implying that the columns of U have been normalized.
4.6. The Inverse of Unit Vector Matrix U
The normal definition of the inverse of a variable is that the inverse times the variable itself yields a unity result. In matrix notation, for our unit vector matrix U, this becomes:
![]() (25)
(25)
The unit matrix Iq is again a diagonal matrix: its non-zero elements are all 1 and are all along the diagonal from the top-left to the lower right of this square matrix. The “left-inverse” of the matrix U is identical to its transposed version UT (see above). We will thus use these as being identical below. We will use for example: (UT)−1 = U.
4.7. Conjugate Representation Spaces
It may already have been assumed implicitly above, but let us emphasise one aspect of the matrix representation explicitly. Each row of the X matrix represents a full image, with all its pixel-values written in one long line. To fix our minds, we introduced a data matrix with 200,000 images (n = 200,000) each containing 90,000 pixel densities (p = 90,000). This data set can thus be seen a data cloud of n points in a p-dimensional “image space”. An alternative hyperspace representation is equally valid, namely, that of a cloud of p points in an n-dimensional hyper space. The co-ordinates in this conjugate n-dimensional space are given by the columns of matrix X rather than its rows.
The columns of matrix X correspond to specific pixel densities throughout the stack of images. Such column-vectors can therefore be called “pixel-vectors”. The first column of matrix X thus corresponds to the top-left pixel density throughout the whole stack of n input images. Associated with the matrix X are two hyper-spaces in which the full data set can be represented: 1) the image-space in which every of the n images is represented as a point. This space has as many dimensions as there are pixels in the image; image-space is thus p-dimensional. The set of n points in this space is called the image-cloud; 2) the pixel-space in which every one of the p pixel-vectors is represented as a single point: pixel-space is n-dimensional. The set of p points in this space is called the pixel-vector-cloud or short: pixel-cloud. (This application-specific nomenclature will obviously change depending on the type of measurements we are processing.)
Note that the pixel vectors are also the rows of the transposed data matrix XT. We could have chosen those vectors as our basic measurements entering the analysis without changing anything. As we will see below, the analyses in both conjugate spaces are fully equivalent and they can be transformed into each other through “transition formulas”. There is no more information in one space than in the other! In the example we chose n = 200,000 and p = 90,000. The fact that n is larger than p means that the intrinsic dimensionally of the data matrix X here is “p”. Had we had fewer images n than pixels p in each image, the intrinsic dimensionally or the rank of X would have been limited to n. The rank of the matrix is the maximum number of possible independent (non-zero) unit vectors needed to span either pixel-vector space or image space.
5. Mathematics of MSA Data Compression
The mathematics of the PCA eigenvector eigenvalue procedures have been described in various places (for example in [14] [15] [33] ). We here try to follow what we consider the best of earlier presentations with a bit of a further personal twist. We want to find a unit vector that best describes the main direction of elongation of the data cloud. “Best” here means finding a direction which best describes the variance of the data cloud.
5.1. MSA: An Optimization Problem
Let direction vector “u” be the vector we are after (Figure 2); the variance of image I that is described by a vector u is the square of the length of the projection of image I onto the vector u, that is![]() . If the vector u is to
. If the vector u is to
maximize the variance it describes of the full data cloud, we need to maximize ![]() where the sum is over all the n images in the data cloud. In doing so, we are also minimizing
where the sum is over all the n images in the data cloud. In doing so, we are also minimizing![]() , the sum of the square distances
, the sum of the square distances
of all images to the vector u, making this a standard least-square minimization problem. We have seen above (Equation (18)) how to calculate the inner product of the full data matrix X and a unit vector u.
![]() (26)
(26)
![]()
Figure 2. Finding the main variance axis of the data cloud. Each image, represented by a single point in a p-dimensional hyperspace, is projected onto the vector u. Together; these points form a data “cloud”. We aim at maximizing the sum of squares of these projections (inner products). The quantity to maximize is thus ![]() where the sum is over all the n images in the data cloud.
where the sum is over all the n images in the data cloud.
The sum ![]() we want to maximize is the inner product of this resulting co-ordinate vector with itself or:
we want to maximize is the inner product of this resulting co-ordinate vector with itself or:
![]() . We thus can write this as the variance we want to maximise:
. We thus can write this as the variance we want to maximise:
![]() (27)
(27)
Let u1 be the unit vector that maximises this variance and let us call that maximised variance λ1. (We will see below how this maximum is actually calculated). We then have for this variance maximizing vector:
![]() (28a)
(28a)
Since u1 is a unit vector we have (see above) the additional normalisation condition:
![]() (28b)
(28b)
The data matrix has many more dimensions (keyword “rank”) than can be covered by just its main “eigenvector” u1, which describes only λ1 of the total variance of the data set. (As mentioned above, the total variance of the data set is the sum of the diagonal elements of A, known as its trace). We want the second eigenvector u2 to optimally describe the variance in the data cloud that has not yet been described by the first one u1. We thus want:
![]() (28c)
(28c)
While, at the same time, u2 is normalized and perpendicular to the first eigenvector, thus:
![]() (28d)
(28d)
and,
![]() (28e)
(28e)
5.2. Eigenvector Equation in Image Space
It now becomes more appropriate to write these “eigenvector eigenvalue” equations in full matrix notation. The matrix U contains eigenvector u1 as its first column, u2 as its second column, etc. The matrix Λ is a diagonal matrix with as its diagonal elements the eigenvalues λ1, λ2, λ3, etc.:
![]() (29a)
(29a)
with the additional orthonormalization condition:
![]() (29b)
(29b)
The eigenvector eigenvalue Equation (29a) is normally written as:
![]() (29c)
(29c)
which is the result of multiplying both sides of Equation (29a) by![]() .
.
5.3. Eigenvector Equation in the Conjugate Pixel-Vector Space
Let the eigenvectors in the space of the columns of the matrix A be called V (with v1 the first eigenvector of the space as its first column, v2 the second column of the matrix V, etc.). The eigenvector equation in this “pixel- vector” space is very similar to the one above (Equation (29a)):
![]() (30a)
(30a)
With the additional orthonormalization condition:
![]() (30b)
(30b)
Both terms of Equation (30a) multiplied from the left by ![]() again yields:
again yields:
![]() (30c)
(30c)
It is obvious that the total variance described in both image space and pixel-vector space is the same since the total sum of the squares of the elements of all row of matrix X is the same as the total sum of the squares of the elements of all columns of matrix X. The intimacy of both representations goes much further, as we see will below.
5.4. Transition Formulas
Multiplying both sides of the eigenvector Equation (29c) from the left with the data matrix X yields:
![]() (31)
(31)
This equation is immediately recognised as the eigenvector equation in the conjugate space of the pixel vectors, Equation (30c) with the product matrix (X∙U) taking the place of the eigenvector matrix V. Similarly, multiplying both sides of the eigenvector equation (30c) from the left with the transposed data matrix XT yields
![]() (32)
(32)
Again, this equation is immediately recognised as the eigenvector equation in image space (Equation (29c)) with the product matrix XT∙V taking the place of the eigenvector matrix U. However, the product matrices X∙U and XT∙V are not normalised the same way as are the eigenvector matrices V and U, respectively. The matrix U, is normalised through UT∙U = Iq (Equation (29b)), but the norm of the corresponding product matrix XT∙V is given by eigenvector Equation (30a):![]() . In order to equate the two we thus need to scale the product matrix by the square root of the eigenvalues:
. In order to equate the two we thus need to scale the product matrix by the square root of the eigenvalues:
![]() (33)
(33)
and, correspondingly:
![]() (34)
(34)
These important formulas are known as the transition formulas relating the eigenvectors in image space (p space) to the eigenvectors in pixel-vector space (n space).
5.5. Co-Ordinate Calculations
We mentioned earlier that the co-ordinates of the images in the space spanned by the unit vectors U are the product of X and U (Equation (23)); we now expand on that, using transition formulas (34) and (33):
![]() (35)
(35)
and their pixel-vector space equivalents
![]() (36)
(36)
5.6. Eigen-Filtering/Reconstitution Formulas
We have seen that the co-ordinates of the images with respect to the eigenvectors (or any other orthogonal co-ordinate system of p space) are given by: X∙U = Cimg (Equation (23)). Multiplying both sides of that equation from the right by UT (a q × p matrix) yields:
![]() (37a)
(37a)
Using Equation (34) we can also write this equation as
![]() (37b)
(37b)
This formula is known as the eigen-filtering or reconstitution formulas [14] [15] as they allow us to recalculate the original data from the co-ordinate matrices and eigenvectors/eigenvalues. Note that the dimensions of V are (n × q) and those of UT are (q × p), with q being maximally the size of n or p (whichever is smaller).
The reason for using a “*” to distinguish X* from the original X, is the following: we are often only interested in the more important eigenvectors, assuming that the higher eigenvectors and eigenvalues are associated with experimental noise rather than with real information we seek to understand. Therefore we may restrict ourselves to a relatively small number of eigenvectors, or restrict ourselves to a value for q, of say, 50. The formulas can now be used to recreate the original data (X) but restricting ourselves to only that information that we consider important.
6. Mathematics of MSA with Generalized Metrics
We have introduced various distances and correlation measures earlier, but in discussing the MSA approaches we have so far only considered conventional Euclidean metrics.
6.1. The Diagonal Metric Matrices N and M
We have discussed above that Euclidean distances are not always the best way to compare measurements and that it may be sometimes better to normalize the measurements by their total (c2 distances) or by their standard deviation (Modulation distance). In matrix notation let us introduce an n × n diagonal weight matrix N that has as its diagonal elements 1/wi, where wi is, say, the average density of image xi, or the standard deviation of that image (row i of the data matrix X). Note that then the new product matrix ![]() = N∙X will have rows
= N∙X will have rows ![]() which will have all its elements divided by the weight wi. We then calculate the associated variance-covariance matrix:
which will have all its elements divided by the weight wi. We then calculate the associated variance-covariance matrix:
![]() (38)
(38)
Interestingly, now all the elements of this variance co-variance matrix are normalized by the specific weights wi for each original image as required for the correlations we discussed above for the c2 metrics (Equation (9)) or the modulation metrics (Equation (13)).
Similarly, we can introduce a diagonal (p × p) weights matrix M in the conjugate space with diagonal elements 1/wj, where wj is, the average density of pixel-vector xj, or the standard deviation of that pixel vector (column j of the data matrix X). Note that then the product matrix X' = X∙M will have columns ![]() which will have all its elements divided by the weight wj. Lets us now combine these weight matrices in both conjugate spaces into a single formulation. Instead of the original data matrix X we would actually like to use a normalized version X' which relates to the original data matrix X as follows:
which will have all its elements divided by the weight wj. Lets us now combine these weight matrices in both conjugate spaces into a single formulation. Instead of the original data matrix X we would actually like to use a normalized version X' which relates to the original data matrix X as follows:
![]() (39)
(39)
and its transposed:
![]() (40)
(40)
6.2. Pretreatment of X with Metric Matrices N and M
Let us now substitute these in to the PCA eigenvector eigenvalue Equation (29c):
![]()
Leading to:
![]() (41)
(41)
with the additional (unchanged) orthonormalization constraint (29b):
![]()
With the N and M normalisations of the data matrix X, nothing really changed with respect to the mathematics of the PCA calculations with Euclidean metrics discussed in the previous paragraphs. All the important formulas can be simply generated by the substitution above (Equation (39)). For example, the co-ordinate Equation (35) becomes:
![]() (42)
(42)
We call this pretreatment because this multiplication of X with N and M can be performed prior to the eigenvector analysis exactly the same way as the pretreatment band-pass filtering of the data discussed above. The procedures of the MSA analysis are not affected by pretreatment of the data (although the results can differ substantially).
The normalisation of the data by N and M allow us to perform the eigenvector analysis from a perspective of c2 distances or that of modulation distances. This normalisation means that, in the 9,000,000 bicycle example for c2 distances, the measurements for Beijing and Cambridge fall on top of each other which is what we wanted. However, the fact that the weight of the measurement for Beijing is 100 times higher than that for Cambridge will be completely lost with this normalisation! That means that even for the calculation of the eigenvectors and eigenvalues of the system, the weight of Cambridge contribution remains identical to that of Beijing.
In standard (not normalised PCA), the contribution of Beijing to the total variance of the data set to the eigenvalue/eigenvector calculations would be 1002 =10,000 times higher than that of Cambridge, thus distorting the statistics data set. (Squared correlation functions in general suffer from this problem [34] ). It was this distortion of the correlation values that prompted the introduction of the normalisation matrices N and M in the first place. However, with the full compensation of the standard deviations of total averages through the N and M matrices we may thus have overdone what we aimed to achieve.
6.3. MSA Formulas with Generalized Metrics in “p Space”
A more balanced approach than either the pure PCA approach or the total normalisation of the data matrix can be achieved by concentrating our efforts on a partially normalised data matrix ![]()
![]() (43a)
(43a)
and its transposed:
![]() (43b)
(43b)
Substituting these in to the classical PCA eigenvector-eigenvalue equation yields:
![]() (44a)
(44a)
![]() (44b)
(44b)
with the additional (unchanged) orthonormalization constraint
![]() (44c)
(44c)
By then substituting ![]() we obtain the eigenvector-eigenvalue equation:
we obtain the eigenvector-eigenvalue equation:
![]() (45)
(45)
This is equivalent to (multiplying left and right hand side of the equation from the left by M−1/2) the eigenvector-eigenvalue equation for generalised metrics [14] [15] [33] :
![]() (46a)
(46a)
However, by substituting![]() , (and equivalently in the conjugate space
, (and equivalently in the conjugate space![]() ) we deliberately choose the co-ordinate system itself to reflect the different weights of the columns and rows of the data matrix and the orthonormalization condition now rather becomes:
) we deliberately choose the co-ordinate system itself to reflect the different weights of the columns and rows of the data matrix and the orthonormalization condition now rather becomes:
![]() (46b)
(46b)
6.4. MSA Basic Formulas with Generalized Metrics in “n Space”
Equivalently, we obtain the eigenvector-eigenvalue equation in the conjugate space as:
![]() (47a)
(47a)
or, alternatively, formulated as (the result of a multiplication from the left with (N−1/2)
![]() (47b)
(47b)
with the associated orthonormalization condition
![]() (47c)
(47c)
6.5. Transition Formulas with Generalized Metrics
For deriving the transition formulas we proceed as was done earlier for PCA derivations. Starting from the eigenvector-eigenvalue Equation (45), and multiplying both sides of this equation from the left with the normalized data matrix ![]() yields:
yields:
![]() (48)
(48)
This last equation, again, is virtually identical to the eigenvector equation in the conjugate space (apart from its scaling):
![]() (49)
(49)
And, again, we have a different normalisation for![]() . The latter has a unity norm (see Equation (47c)), whereas
. The latter has a unity norm (see Equation (47c)), whereas ![]() has the norm Λ as becomes clear from multiplying Equation (45) from the left with
has the norm Λ as becomes clear from multiplying Equation (45) from the left with ![]() yielding:
yielding:
![]() (50)
(50)
We thus again need to normalise the “transition equation” with Λ−1/2, leading to two transition equations between both conjugate spaces:
![]() (51)
(51)
and correspondingly:
![]() (52)
(52)
6.6. Calculating Co-Ordinates with Generalized Metrics
The calculation of the image co-ordinates in n space as we have seen above (Equation (23)):
![]() (23’)
(23’)
With the appropriate substitutions:
![]() (53)
(53)
and
![]() (54)
(54)
However, these co-ordinates, seen with respect to the eigenvectors U, have a problem: the matrix ![]() is only partially normalised with respect to N. With the example of Beijing versus the Cambridge bicycle density, Beijing has a hundred times higher co-ordinate values than Cambridge, while having exactly the same profile. For the generalised metric MSA we thus rather use the co-ordinates normalised fully by N and not just by N1/2 [14] [15] [33] :
is only partially normalised with respect to N. With the example of Beijing versus the Cambridge bicycle density, Beijing has a hundred times higher co-ordinate values than Cambridge, while having exactly the same profile. For the generalised metric MSA we thus rather use the co-ordinates normalised fully by N and not just by N1/2 [14] [15] [33] :
![]() (55)
(55)
(The right hand side was derived using the transition formula ![]() (Equation (51)) multiplied from the right by Λ1/2.) And we also have, similarly:
(Equation (51)) multiplied from the right by Λ1/2.) And we also have, similarly:
![]() (56)
(56)
Using these co-ordinates for the compressed data space again puts the “Beijing measurement” smack on top of the “Cambridge measurement”. How is this now different from the “total normalisation” discussed in section 6b (above)? The difference lies in that each measurement is now not only associated with its co-ordinates with respect to the main eigenvectors of the data cloud, but each measurement now is also associated with a weight. The weight for the “Beijing measurement” here is one hundred times higher than that of the “Cambridge measurement”. That weight difference is later taken into account, for example, when performing an automatic hierarchical classification of the data in the compressed eigenvector space.
7. MSA: An Iterative Eigenvector/Eigenvalue
The algorithm we use for finding the main eigenvectors and eigenvalues of the data cloud is itself illustrative for the whole data compression operation. The IMAGIC “MSA” program, originally written by one of us (MvH) in the early 1980s, is optimised for efficiently finding the predominant eigenvectors/eigenvalues of extremely large sets of images. Here we give a simplified version of the underlying mathematics. Excluded from the mathematics presented here are the “metric” matrices N and M for didactical reasons. The basic principle of the MSA algorithm is the old and relatively simple “power” procedure (cf. [35] ; also discussed in Wikipedia under “eigenvector power iteration”). In this traditional approach one multiplies a randomly chosen vector r1, through the symmetric variance co-variance matrix A, which will yield a new vector![]() :
:
![]() (57a)
(57a)
This resulting vector is then (after normalisation) successively multiplied through the matrix A again:
![]() (57b)
(57b)
and that procedure is then repeated iteratively. The resulting vector will gradually converge towards the first (largest) eigenvector u1 of the system, for which, per definition, the following equation holds:
![]() (58)
(58)
Why do these iterative multiplications necessarily iterate towards the largest eigenvector of the system? The reason is that the eigenvectors “u” form a basis of the n-dimensional data space and that means that our random vector r1 can be expressed as a linear combination of the eigenvectors:
![]() (59)
(59)
The iterative multiplication through the variance-covariance matrix A will yield for r1 after k iterations (using Equation (58) repeatedly):
![]() (60a)
(60a)
or:
![]() (60b)
(60b)
Because λ1 is the predominant eigenvalue, the contributions of the other terms will rapidly vanish (![]() ; i > 1), and these iterations will thus make r1 rapidly converge towards the main eigenvector u1. The variance co-variance matrix A is normally calculated as the matrix multiplication of the data matrix X and it's transposed, XT:
; i > 1), and these iterations will thus make r1 rapidly converge towards the main eigenvector u1. The variance co-variance matrix A is normally calculated as the matrix multiplication of the data matrix X and it's transposed, XT:
![]() (61)
(61)
As was mentioned above, the data matrix X contains, as its first row, all of the pixels of image 1; its general ith row contains all the pixels of image i. The MSA algorithm operates by multiplying a set of randomly generated eigenvectors (because of the nature of the data also called eigenimages) r1, r2, etc., through the data matrix U and its transposed U' respectively. The variance-covariance matrix Ap is thus never calculated explicitly since that operation is already too expensive in terms of its massive computational burden. The MSA algorithm does not use only one random starting vector for the iterations, but rather uses the full set of q eigenimages desired and multiplies that iteratively through the data matrix X, similar to what was suggested by [36] .
In detail the MSA algorithm works as follows (Figure 3). The eigenvector matrix Uq is first filled with random numbers which are then orthonormalised (normalised and made orthogonal to each other). The typical number of eigenimages used depends fully on the complexity of the problem at hand but typically is 10 - 100 and they are symbolised by a set of two “eigenimages” in the illustration (top of Figure 3). Then, the inner product between these images and the n images of the input data set is calculated. This calculation leads to coefficient vectors of length n as indicated in the left-hand side of Figure 3. The next step is then to calculate weighted sums over the input image stack, using the different coefficient vectors as the weights for the summing. A new set of eigenimage approximations is thus generated as shown in the lower part of Figure 3. New approximations are generated from this set by orthonormalization and over-relaxation with respect to the previous
![]()
Figure 3. The iterative MSA eigenvector-eigenvalue algorithm. The program first fills a matrix U with orthonormalised random column number vectors (E1', E2', etc.). This matrix is then multiplied through the data matrix X containing the set of images I1 through In as rows. This multiplication leads to a matrix of coefficients C which is then multiplied through the transposed data matrix XT leading to (after normalization) a better approximation of the eigenvector matrix. In essence the algorithm performs a continuous set of iterations through the eigenvector Equation (29a)![]() . (For the equivalent including the influence of generalized metrics, we have from Equation (46a):
. (For the equivalent including the influence of generalized metrics, we have from Equation (46a):![]() ).
).
set. The algorithm converges rapidly (typically within 30 - 50 iterations) to the most important eigenimages of the data set.
An important property of this algorithm is its efficiency for large numbers of images n: its computational requirements scale proportionally to n∙p, assuming the number of active pixels in each image to be p. Most eigenvector-eigenvalue algorithms require the variance-covariance matrix as input. The calculation of the variance-covariance matrix, however, is itself a computationally expensive algorithm requiring computational resources almost proportional to n3. (This number is actually: Min (n2p, np2)). The MSA program produces both the eigenimages and the associated eigenpixel-vectors in the conjugate data space as described in [33] . One of the intuitive charms of this fast disk-based eigenvector-eigenvalue algorithm is that it literally sifts through the image information while finding the main eigenimages of the data set. The programs have been used routinely for more than 30 years, on a large number data sets consisting of up to ~1,000,000 individual images.
8. Parallelization of the MSA Algorithm
In spite of its high efficiency and perfect scaling with the continuously expanding sizes of the data sets, compared to most conventional eigenvector-eigenvalue algorithms, the single-CPU version of the algorithm had become a serious bottleneck for the processing of large cryo-EM data sets. The parallelisation of the MSA algorithm had thus moved to the top of our priority list. We have considered various parallelisation schemes including the one depicted in Figure 4; this mapping of the computational problem onto a cluster of computers was indeed found to be efficient.
As was discussed before, Equation (46a) (see also the legend of Figure 3) is the full matrix equivalent of:
![]()
Figure 4. Parallelisation of the MSA algorithm. Note that for an efficient operation it is essential that the part of the input data matrix X that is of relevance to one particular node of the cluster, is indeed always available on a high-speed local disk.
![]() ;
;![]() (62)
(62)
In which, uj is one single eigenvector and λj the corresponding eigenvalue. Having stated this, a direct approach for parallelisation of the calculation of Equation (46a) could be the exploiting of the independence of calculations of all uj since each eigenvector ![]() depends only on one eigenvector uj. Although it is a clear possibility and is straightforward to implement, this scheme does not represent an effective approach. Since there may typically be more processors available than the number of eigenvectors needed for representing the dataset, this approach would lead to a waste of processing time with many processors runningidle. Moreover, all processors would need access to the full image matrix X. This approach is thus likely to create severe I/O bottlenecks. To analyse another possible parallelisation schemes, let us write Equation (62) as:
depends only on one eigenvector uj. Although it is a clear possibility and is straightforward to implement, this scheme does not represent an effective approach. Since there may typically be more processors available than the number of eigenvectors needed for representing the dataset, this approach would lead to a waste of processing time with many processors runningidle. Moreover, all processors would need access to the full image matrix X. This approach is thus likely to create severe I/O bottlenecks. To analyse another possible parallelisation schemes, let us write Equation (62) as:
![]() (63)
(63)
where uij is element i of eigenvector uj, xij is pixel j of image i, and ns and mt are the diagonal elements of the N and M metrics, respectively. This equation can be expanded into the form:
![]() (64)
(64)
The equation above suggests a possible parallelisation scheme over the image dataset instead of the eigenvectors (Figure 4). Each processor calculates a partial solution for ![]() based on a subset of k images. After the complete calculation of U', the further steps of the algorithm are computed in a non-parallel way. The algorithm thus consists of a parallel and a non-parallel part.
based on a subset of k images. After the complete calculation of U', the further steps of the algorithm are computed in a non-parallel way. The algorithm thus consists of a parallel and a non-parallel part.
The implementation of the algorithm can be structured in a master/slave architecture, where the master is responsible for all non-parallel tasks and for summing the partial results of each node. During each iteration of the algorithm, each node needs to read data from the image matrix X thus potentially creating a network bottleneck. However, since each node in this parallelisation scheme needs only to access a part of the X matrix, distributing the partial datasets of input data over the local disks efficiently, parallelises the I/O operations.
9. Exploiting the Compressed Information in Factor Space
What we wish to achieve with the MSA approaches is to concentrate on the intrinsic information of large noisy data sets and make that more accessible. The determination of the eigenvectors (eigenimages) by itself does not change or reduce the total image information in any way. It is merely a rotation of the co-ordinate system in a special direction, such that the first eigenvector covers most of the variance in the data cloud, the second, orthogonal to the first, covers most of the remaining variance of the data cloud, not covered by the first eigenvector, etc. If one would find all the eigenvectors of the data cloud (all the eigenvectors of the variance-covariance matrix; all p or n of them, whichever is the smaller number) there would be no loss of information at all. Of course, even without any loss of information the most important information is associated with the first eigenvectors/eigenimages that describe the strongest variations (in terms of variance) within the data set.
When we decide to consider only the first so-many eigenvectors and thus decide to ignore all higher eigenvectors of the system, we make the “political” decision between what is signal and what is noise. Here the decision that the percentage of the overall variance of the data set (the “trace”) covered by the main eigenvectors, suffices for our desired level of understanding of the dataset. An often spectacular level of data compression is thus achieved. The problem remains of how to best exploit that information in the compressed data space. Having concentrated on the mathematics and algorithmic aspects of the eigenvector-eigenvalue aspect of the MSA approach, we here will just give a relatively short account of this most fascinating aspect of the methodology.
10. Visual Classification in a Low-Dimensional Factor Space
The first EM data sets that were ever subjected to the MSA approach were simple negative-stain contrasted specimens where the differences were directly visible in the micrographs. The data sets were chosen carefully for being simple in order for the problem to be solvable with the limited computing resources available in those days. The very first data set that was ever analysed by the MSA approach was an artificial mixture dataset from two different species of hemoglobins. The first analyses were performed with the “AFC” program developed by Jean Pierre Bretaudière from a program originally written by JP Benzécri (see Appendix for details). The 16 molecular images of the giant annelid hemoglobin of Oenone fulgida visibly had extra density in the centre compared to the 16 molecular images of well-known giant hemoglobin of the common earthworm Lumbricus terrestris which rather had a “hole” in the centre of its double-ring geometry (see Appendix). The separation of the two species was so obvious that one immediately moved on to a single data set with real internal variations (the example below).
In those early days in computing, the address space of the computers was minimal by today’s standard. The 16-bit PDP-11/45 computer on which the work was performed would only address 216 bytes = 64 Kbytes of memory. It was thus difficult to fit any serious computational problem in the central memory of that generation of computers! The critical memory requirement for the eigenvector-eigenvalue calculations was the “core” needed for the square variance-covariance matrix which, practically limiting the number of “particles” to around 50 at most. In the IMAGIC implementation of Bretaudière’s AFC program (see Appendix) the limit was slightly over 100 molecular images (see the Limulus polyphemus hemocyanin double-hexamer analysis in [39] ). That larger number of particles was purely due to the fact that the NORSK DATA Nord-10 computer addressed 16-bit word rather than the 8-bit words of the PDP-11/45, a doubling of the usable memory. With the computer hardware limiting the complexity of problems that could be reasonably studied, the limitations of being able to only look at problems that were intrinsically two-dimensional, was not immediately felt. It was more frustrating that the averaging was over classes that still were a subjective choice of the user after the elegant data compression.
11. MSA Automatic Classification Algorithms
To eliminate the subjectivity of visual classification (which is simply impossible when the number of factors to be taken into account is larger than two or three), automatic classification schemes were introduced in EM. These schemes emerged in the general statistical literature in the 1960s and 1970s, and were first used in electron microscopy in the early 1980s. There are many algorithms available each with their advantages and disadvantages. The three listed below are important in single particle cryo-EM.
11.1. K-Means Classification Algorithms
In the first class of algorithms, one chooses a number of k classes into which one wishes to use to subdivide the full data set (of n images) and then selects at random k from the n elements to serve as the first classification
![]()
Figure 5. Visual Classification. Historical “correspondence analysis” of negatively stained “4 × 6” half-molecules of the hemocyanin of Limulus polyphemus. The two double-hexameric rods constituting this molecule are shifted with respect to each other thus forming a parallelepiped which looks different in face-up and face-down position (“flip” and “flop”). The four hexamers constituting the 4 × 6-mer were, however, apparently not in a single plane, which leads to two stable “rocking” positions where 3 out of 4 hexamers are in touch with the carbon support film, and one not and is therefore stained less. This effect splits both the “flip” and the “flop” groups in two “rocking” positions. This analysis was performed with an early version of the IMAGIC software [37] ; the illustration is reproduced from [38] .
11.2. Hierarchical Ascendant Classification (HAC)
The HAC technique is a “from-the-bottom-up” classification approach in which each element of the data set is first considered as a “class” by itself. Each of these individual starting classes can be associated with a mass depending on the choice of metric. Individual classes are then merged in pairs, based on a merging criterion, until finally one single large class emerges, containing all the elements of the original data set. The history of the classification procedure (that is, which classes merge together at which value of the aggregation criterion) is stored in a classification or merging “tree”. The user chooses the desired number of classes at which the tree is to be cut. As a merging criterion, the minimum added intra-class variance or Ward criterion is normally used [13] [15] [43] . The algorithm is aimed at minimising the internal variance of the classes (“intra-class variance”) while at the same time maximising the inter-class variance between the centres of mass of the class averages. At any level of the procedure, two classes are merged to form a new, larger class if the increase in total intra-class variance associated with their merging is the lowest possible at that level. The Added-Intra-class-Variance associated with the merging of class i with class j is:
![]() (65)
(65)
In this formula ![]() is the (now) Euclidean square distance between the classes i and j having masses (weights) wi and wj respectively (Appendix of [16] ). To obtain a predefined number of classes from the process, one then cuts the merging “tree” at the appropriate level. The advantage of this type of algorithm is that it provides a logical and consistent procedure to subdivide the data set into various numbers of classes, whatever number makes sense in terms of the specific problem at hand. The algorithm helps visualising the inherent structure of the data by showing―at all levels―which classes are to merge with which classes. The first use of HAC schemes in electron microscopy was in [16] [44] . Because of the explicit influence of the masses in HAC (Equation (65)) the approach is well suited for the generalised metrics including the c2-metric and the modulation metric.
is the (now) Euclidean square distance between the classes i and j having masses (weights) wi and wj respectively (Appendix of [16] ). To obtain a predefined number of classes from the process, one then cuts the merging “tree” at the appropriate level. The advantage of this type of algorithm is that it provides a logical and consistent procedure to subdivide the data set into various numbers of classes, whatever number makes sense in terms of the specific problem at hand. The algorithm helps visualising the inherent structure of the data by showing―at all levels―which classes are to merge with which classes. The first use of HAC schemes in electron microscopy was in [16] [44] . Because of the explicit influence of the masses in HAC (Equation (65)) the approach is well suited for the generalised metrics including the c2-metric and the modulation metric.
A disadvantage of the HAC algorithm [16] [17] [33] compared to the k-means schemes, however, are the computational efforts required. For smaller data sets of up to ~10,000 images/elements, the computational requirements are negligible compared to those of the eigenvector-eigenvalue calculations themselves. However, since the computational requirements of the algorithm grow proportionally to ~n2, (with n the number of images in data set) for larger data sets of, say, more than 200,000 elements, the computational requirements become excessive and often exceed the requirements for the eigenvector calculations.
Although, at every merging step, two classes are merged that lead to a minimum AIV contribution, this fact is also a fundamental limitation: If two elements are merged into one class at an early stage of the procedure, the elements will always remain together throughout all further HAC classification levels, whereas if their marital ties were weaker, a lower intra-class variance minimum could easily be obtained [17] . Also, the merging of two classes at a later level of the HAC algorithm may locally cause a large increase in intra-class variance. A simple yet most effective post-processor was designed to deal with the problem that any HAC partition is typically far from a local minimum of the total intra-class variance.
11.3. Moving Elements Refinement
The moving-elements approach is a post-processor to refine and consolidate an existing partition. Starting point is typically the partition obtained with HAC based on the Ward criterion discussed above. The partition is refined, to reach a “deeper” local minimum of intra-class variance, by allowing each member of each class to migrate to any other class where it is happier in terms of the very same added intra-class variance criterion [17] [30] . Using the HAC partition as a starting point, each element is extracted from its HAC class and the AIV distances relative to all other classes are calculated for that particular element. If the minimum of these AIV values is lower than the element’s AIV distance to its current class, it is extracted from its current class and placed into the other one. The statistics of both its old and its new class are updated and the algorithm proceeds with the next element.
After one cycle, many of the classes have changed and the procedure is iterated until no (or little) further moving of elements is observed. The partition thus obtained is a real (and significantly deeper) local minimum of the total intra-class variance than that obtained directly by the HAC in the sense that no single element can change class-membership without increasing the total intra-class variance of the partition. We call this algorithm “moving elements consolidation” (or refinement) as opposed to the “moving centres consolidation” proposed independently by [45] , which is a pure k-means post-processor to the HAC partition based on the same basic idea that HAC partitions are prone to improvement. The HAC scheme, in combination with the moving elements post-processor, has emerged as the most robust classification scheme.
11.4. Hybrid Classification Refinements
A variety of further possible classification schemes/refinement schemes have been have suggested in [17] and we refer the reader to that paper directly. Here it suffices to state that some of those schemes have since been implemented and are used in our daily routine. We have chosen to primarily work with the Ward criterion discussed above. In particular, the “hybrid classification” implemented in the recent versions of the IMAGIC-4D software [46] operate along the following lines. A) From the large number of input measurements, say 1,000,000 images, we first choose a set of, say 20,000 random seeds. These seeds are declared “mergeable” implying that all images are only compared to the seed images and that the HAC merging operations of original images are restricted to classes containing the starting seeds. Thus, instead of dealing with 1,000,0002 comparisons we only have to deal with 1,000,000 × 20,000 comparisons. The HAC operations are otherwise fully standard and it stops when the desired final number of classes is reached (say, 2000). The moving elements post-processor, again still based on the Ward criterion, bring the whole partition to a variance minimum.
12. MSA Symmetry Analysis
Another example of the use of MSA is the unbiased analysis of the main symmetry properties of a macromolecular complex. Earlier symmetry analysis approaches were based on finding the predominant rotational symmetry components of one single image at a time [49] . That single image could then be used to align all particles of the data set. However, when aligning a whole set of images with respect to one single image with a strong, say, 6-fold symmetry component, the 6-fold symmetry property will be implicitly imposed upon the whole data set by the reference-bias effect [50] [51] during the alignment procedure. When an average is then obtained from the aligned images, the 6-fold symmetry component becomes overwhelmingly present, but that serves only as a demonstration of the reference bias effect, and certainly not as a proof of the real symmetry of the complex being studied. Various papers have appeared in the literature in which this “self-fulfilling prophecy” approach was used for determining the “true” symmetry of an oligomeric biological structure.
A methodologically clean approach for determining the strongest symmetry component in a data was proposed, which entirely avoids the symmetry bias resulting from any explicit or implicit rotational alignment of the molecular images [52] . In this approach, only a translational alignment relative to a rotationally symmetric “blob” is performed for centring particles. The rotational orientation of all molecules remains arbitrary, and will thus often be the main source of variation among the images of the data set. MSA eigenvector analysis will then find those main eigenimages of the system and these reflect the symmetry properties of data elegantly. The particles used here (Figure 6) resulted from an automatic particle selection over a stack of micrographs of the hemoglobin of the common earthworm Lumbricus terrestris. We use these data as a high-contrast general testing standard [53] . The particle picking program was used in cross-correlation mode with a rotationally-symmetric reference/template image (see insert in Figure 6). This particle-particle picking procedure yields centred particles equivalent to the translational alignment with a rotationally symmetric average used in the original paper [52] .
The total of some 7300 particles were submitted to MSA eigenvector analysis. The first eigenvector in this analysis (Figure 7) is almost identical to the reference image used to pick the particles. The two next eigenimages are essentially two identical 6-fold symmetrical images rotated by a small angle with respect to each other. A six-fold symmetrical structure repeats itself after a rotation over 60˚. These two eigenimages 2 and 3, are rotated by exactly 15˚ degrees with respect to each other. The eigenimages 2 and 3 consist of sine waves along the tangential direction of the images and along each circle around the centre of the eigenimage we will see 6 full periods of that sine wave. One period of the sine wave corresponds to a 60˚ arc on the circle. The 15˚ rotation between the two eigenimages mean that the sine waves at each radius of the two images are π/2 out of phase with each other, like a sine and a cosine function. Note also (from the eigenvalue plot in Figure 7) that they have an associated eigenvalue of more than twice the magnitude of all subsequent eigenvalues.
![]()
Figure 6. Symmetry analysis. Some of the ~7300 particles selected automatically from a set of micrographs using the automatic particle picking program PICK_M_ALL (part of the IMAGIC-4D software system [46] ). The rotationally symmetric template image shown in the top-left corner of this illustration was used to find the particles by cross correlation. This procedure yields particles that are centred with respect to the reference image. These particles have no bias to any reference image in a rotational direction and such a data set is ideal input for subsequent MSA-based symmetry analysis (Figure 7).
![]()
Figure 7. Main eigenimages. The first 25 eigenimages of the data set of ~7300 particles (Figure 6), together with a plot of the associated 10 first eigenvalues. Eigenimages 2 and 3 exhibit perfect 6-fold symmetry, and are virtually identical to each other apart from a small rotation. They are rotated by exactly 15˚ degrees with respect to each other meaning that the tangential sine waves at each radius of the two images are π/2 (90˚) out of phase with each other. Note from the eigenvalue plot, that the first eigenvector covers ~2% of the total variance of the data set, and that the first 10 eigenvectors together cover around 4.5% of that total variance.
13. Alignments and MSA
Alignments of the images within the data matrix X change the MSA analysis in fundamental ways. Interestingly however, alignments of the individual images do not change the total variance of the data set. A rotation of an image merely shifts around the pixel densities within a row of the data matrix X; the total variance of the measurement, however, does not change. (This is perfectly true as long as no non-zero pixels are rotated or shifted out of the part of the image that is active during the MSA analysis). The variance of each image is the corresponding diagonal element of the variance-covariance matrix A and hence these diagonal elements remain unchanged.
The total variance of the data set (the trace of A) is the sum of the diagonal elements of matrix A (Equation (21a)). What does change during the alignment procedures is that certain images within the data matrix will become better aligned to each other and will sense a higher co-variance among themselves (specific off-diagonal elements of matrix “A” will increase).
We will here apply an interesting and illustrative form of alignment, namely: “alignment by MSA”, (similar to the Alignment By Classification “ABC” [46] [52] ) to the worm hemoglobin data set introduced above (Figure 6). Having noticed that eigenvectors 2 - 7, all are associated with 6-fold symmetry or the harmonics thereof, we choose to align the (already centred) data set-in a rotational sense only-with respect to eigenimage 2 (Figure 8). The rotation alignment was restricted in range of −30˚ to +30˚ in order to leave the 6-fold-symmetry character of the data set undisturbed. Eigenimage 2 is pasted in the top-left corner of the illustration, whereas all the other images in this figure are the rotationally aligned versions of the centred particle of the original data set (Figure 6).
We have here aligned the full (centred) data set rotationally with respect to one of the two main eigenimages associated with the 6-fold symmetry property of this hemoglobin. In doing so, we have concentrated a much larger percentage of the total variance of the data set into the first few eigenvectors of the system as is seen from the total sum of the first 10 eigenvalues before (4.3%; Figure 7) and after the rotation-only alignment (6.6%; Figure 9). As was mentioned above, an alignment does not change the variance of each individual image and thus does not change the total variance of the data set.
Concentrating the information (variance) into the first few eigenimages means that more eigenvectors will become statistically significant above the background random noise level. With the relatively small data sets from the past, alignments were an absolute necessity for obtaining statistically significant results. Today, however,
![]()
Figure 8. Alignment to eigenimage. Some of the ~7300 automatically selected particles introduced above (Figure 6), but now ro-tated using the second eigenimage as a reference for a rotation-only alignment. The rotation was restricted to −30˚ to +30˚ to stay within the 6-fold symmetry properties of the data set. The now rotationally aligned data set was again submitted to MSA eigenvector analysis; the results of that analysis are shown in Figure 9.
![]()
Figure 9. Eigenimages after alignment to eigenimage #2. MSA eigenvector analysis of the centred data set, but now aligned rotationally with respect to the second eigenimage of the first MSA analysis (Figure 7). The first eigenvector now is clearly recognisable as a top view of the Lumbricus terrestris hemoglobin. It alone now covers ~4.3% of the total variance of the data set (compared to the 2.01% the first eigenvector covered in the first analysis (Figure 7). The first 10 eigenvectors together now cover around 6.6% of the total variance of the data set.
and especially with the powerful new parallel MSA algorithms, we can afford to largely increase the overall size of the data set. The rationale for using larger data sets is simple: using ten times more images in a data set means using ten times more information! In doing so, we can achieve statistical significance without the risk prejudicing the data sets by introducing reference bias [50] [51] . This increased level of objectivity is especially important when trying to find objectively find different conformations in a heterogeneous population of molecular images.
14. MSA and Heterogeneous Data Sets: Manifold Separation
Mixed populations of molecular images were originally the curse of single-particle electron microscopy. Originally, one would try to work with an as homogeneous as possible population of macromolecules. The basic assumption for single particle cryo-EM was that all molecules were the same, apart from the fact that each molecule could exhibit a different orientation and be in a different position in the sample. When the data were heterogeneous or flexible, and that heterogeneity is not accounted for, that results in a deterioration of the quality of the final 3D reconstruction since one will then be averaging “cows” and “horses” into the same 3D map. When, on the other hand, one knows how to separate the molecular images into structurally homogeneous subgroups, a whole new world of possibilities opens. One can then study a mixture of closely related structures and from that commence to understand the biological reasons for the distinct 3D structures found in a sample. The name “4D” cryo-EM has been used in this context.
One of the first examples of the challenging possibilities came from a cryo-EM sample of the E. coli 70s ribosome, in complex with Release Factor 3 [54] . It was found the structure simply did not converge towards a high-resolution structure no matter how much effort was invested. However, once the data set was allowed to refine (by multi-reference alignment) towards different 3D reconstructions simultaneously, the sample turned out to consist of a mixture of ribosomes in two different conformational states. The ribosomal complexes were found to co-exist in the pre-translocation and post-translocational states of the 70S ribosome. The “RF3” was also found in two different conformations depending on the conformation of the 70S ribosome itself. Once the data set had been separated into conformational subsets, by local MSA of specific projection images, each subset of images led to a separate 3D structure exhibiting a much higher resolution than the resolution on the overall 3D reconstruction. A further example, where size variations among the particles was found to be a limiting factor, is given by [55] .
Another possibility to study 3D structural variation is to generate a large number of 3D reconstructions and to study those by means of MSA the way 2D molecular images were studied above [56] . The IMAGIC system has been optimised to manage 3D volumes with the same ease and transparency as the system handles sets of 2D images [46] . This has greatly facilitated handling these huge intricate datasets.
A quality criterion for the 4D MSA types of analysis is the resolution achieved in the subset 3D reconstructions. As usual the resolution is quantified by Fourier Shell Correlation (“FSC”, [57] [58] ). There are two conflicting issues here. It is generally true that larger datasets lead to higher resolution results than smaller datasets. An increase of resolution, while decreasing the number of raw images entering in each specific 3D reconstruction, is an important indication for the population of images indeed to belong to different structural subsets. The separation of large data sets into structurally homogeneous subsets must be seen as a major challenge in current biology and this area is in constant flow (see below). We do not want to go into much detail here but rather point the reader to a general review [59] .
15. Discussion
The main goal of the biological sciences is to make sense of the massive and very noisy “data” in terms of elucidating the underlying general principles. No better example than the data sets collected in cryo-EM which are probably among the largest and noisiest currently being collected in the biomedical sciences. Over the past three decades, multivariate statistical analysis approaches have been very successful in helping us sort out complex EM data sets in many different ways, and examples have been discussed such as a first “manifold” separation into two functional states [54] . However, we have not yet seen the full potential of the MSA approaches exploited due the costs of the computing effort. This high cost has hampered the use and the development of potentially very useful further MSA approaches such as the “alignment by MSA” discussed above.
How can that be that the computational effort was simply not affordable? After all, one of the most dramatic developments in science over the past decades, has been the increase in speed in general computing. In spite of massive computing resources becoming available, however, we have seen an even more rapid increase in the size and complexity of the EM data sets. The size of the data sets have been limited not by what one would wish to analyse, but rather by what was practical to analyse. The high-resolution scanning of (analogue) micrographs into digital form was a tedious activity taking up to hours of labour for a single micrograph. With automatic 24/7 data collection directly on the microscope―using direct electron detector 4 k × 4 k cameras [25] [26] ―the primary data-collection bottleneck has been removed. Therefore the speed of the MSA eigenvector computations had become the new bottleneck limiting its use in cryo-EM. The recent parallelisation of MSA programs (operating in the context of the IMAGIC software system [46] [60] ) opens a whole range of new possibilities by speeding up the process by orders of magnitude, exploiting the power of hundreds of CPUs simultaneously.
There are other reasons why the MSA have not always received the appropriate attention, including ignorance. The MSA approaches are not the simplest of techniques to understand and to get acquainted to for the potential biological user. The learning curve is steep and users tend to avoid investing the time necessary to understand the approach. More seriously, however, is that many software systems in use in cryo-EM do not have MSA options, and those that have typically apply standard libraries for performing the analysis. These standard eigenvector-eigenvalue routines normally calculate all eigenvectors and eigenvalues of the system, and those are algorithms that scale proportional to N2, implying that the analysis of data sets of more than a few thousand elements will easily exceed the memory/computing capacity of any modern computer. With the poor scaling of the algorithms it can be very difficult to analyse large data sets, a fact that is also not appreciated in the general statistical literature where data sets are small by cryo-EM standards. Even in the special EM literature this issue is not always appreciated. (See, for example, paragraph 2.5 of [37] where the reasoning is based on the availability of the variance-covariance matrix A, which is already too expensive to calculate as was discussed above). Therefore, in times where datasets of hundreds of thousands or millions of images are the rule, one sees MSA applications often limited to just a few thousand images.
Another issue that has largely been ignored in the literature is that the original c2-metric of correspondence analysis is inappropriate for electron-microscopical phase-contrast data. Historically it happened to be the first MSA technique used because it was available (see Appendix). However, the c2-metric correspondence analysis uses is designed for positive data (counting occurrences in histograms). To make the EM phase-contrast data positive one could threshold the image data to positive values only. This thresholding, however, is not justifiable for general signals and―at the very least―leads to wasting of half of the information for zero-average-density measurements. Alternatively, the negative values can be rendered positive by adding a constant to the data. This too has far-reaching consequences. The strong negative densities will end up as small positive densities and will have very little contribution to the total variance of the data set, whereas the large positive values will become very large positive values with a disproportionally strong contribution to the total variance. For high-resolution EM work, however, the c2-metric must be considered obsolete since the publication of [33] . The metrics that are permissible for general signal processing are the Euclidean distance of principal components analysis and the modulation distance of modulation analysis discussed above.
In recent years, another multivariate statistical approach was introduced to single particle cryo-EM known as the “maximum likelihood” [61] , an “explanatory factor analysis” technique. This approach had a significant number of successes in refining the structure of large complexes [29] [62] . A discussion of the similarities and differences of the two general approaches (“exploratory factor analysis” versus “principal component analysis”) can be found in Wikipedia from which we quote: “In factor analysis, the researcher makes the assumption that an underlying causal model exists, whereas PCA is simply a variable reduction technique”. And: “If the factor model is incorrectly formulated or the assumptions are not met, then factor analysis will give erroneous results. Factor analysis has been used successfully where adequate understanding of the system permits good initial model formulations. Principal component analysis employs a mathematical transformation to the original data with no assumptions about the form of the covariance matrix.” It is still early days in single-particle cryo-EM and in depth comparisons will be performed in the future. The maximum likelihood approaches are typically used in conjunction with reference-based alignments or projection-matching approaches which, are not reference free and are thus in principle are prone to reference bias. The original MSA approaches were preceded by multi-reference alignments [18] [30] which themselves are also not reference free. However, the Euler orientation assignments were performed separately after a separate unsupervised classification which makes the approach less prone to reference bias. The ABC approaches discussed above [46] [52] introduce a new level of reference independence.
The issue of reference bias is of fundamental importance in the cryo-EM field. There has been a much publicised case where two different 3D structure were presented by Mao & Sodrosky and co-workers [63] [64] which were seriously criticized in the field [65] - [67] . By using “supervised classification”, that is, looking for specific molecular projections in noisy micrographs, these authors found what they were looking for to begin with. Their critics accused the authors of looking for “Einstein in random noise” the classical reference-bias experiment in cryo-EM. Interestingly, the unbiased ABC approach (unsupervised classification; no reference images) was used in [65] to refute the results of those authors. Because our eigenvector-eigenvalue data compression approach is simply a “variable reduction technique” is perfectly suited for this type of neutral, unbiased analysis.
New cryo-EM developments allowing the separation of subsets of 3D structures are emerging that have the potential to change the field of structural biology. Atomic resolution structures (~3Å) had hitherto been elucidated mainly by X-ray crystallography, where the biological molecules are confined to the rigidity of a crystal. In X-ray crystallography one collects diffraction amplitudes data that per definition has been averaged over all unit cells of the crystal. For a better understanding of a biological process, however, it is essential to see the sequence of conformational changes and interactions a molecule or complex undergoes during its functional cycle. A sequence of 3D structures or “4D data analysis” is required for this level of understanding. We want to separate the different manifolds in hyperspace, representing the different states the complexes we study can assume. Single-particle cryo-EM provides a direct window into the solution, revealing different views of different complexes, in different structural/functional states [54] [68] [69] . The challenge is to extract all information from the noisy molecular images and to bring the existing structural differences to statistical significance. Revealing the full biological complexity of the data produced by 4D cryo-EM at atomic resolution remains one of the major challenges in modern structural biology. The MSA approaches discussed in this paper are among the best tools for creating a clear picture of what is happening in the solution.
16. Conclusion
New developments in MSA eigenvector-eigenvalue data-compression approaches are expanding the possibilities of single-particle cryo-EM. Decades after its first introduction to electron microscopy, MSA approaches are more alive than ever and they are being tailored for many new tasks. With the orders-of-magnitude speed increase achieved by the MPI parallelisation of the algorithms, huge datasets of Terabytes in size can processed. A rapid expansion of the multivariate statistical techniques has taken place in single-particle cryogenic electron microscopy. One of the most challenging uses of the parallel MSA algorithm is the separation of heterogeneous data sets into different 3D structures associated with the different functional states of the macromolecular complexes we seek to understand. The different functional states of the molecules we study may be better visualised and understood by assessing the different manifolds one finds in the eigenvector compressed dataspace. Thanks to the maturation and proliferation of multivariate statistical data analysis approaches, single-particle electron microscopy techniques are now one of the preferred instruments of structural biology.
Acknowledgements
First let us again point to the first MSA ideas that were inspired by Jean-Pierre Bretaudière (see Appendix). This document reflects decades of development and experience with the MSA techniques discussed. During this period many individuals contributed to the various MSA projects (and not just the current authors) including: George Harauz, Lisa Borland, Ralf Schmidt, Elena Orlova, Holger Stark, HansPeter Winkler, Manfred Kastowsky, Bruno Klaholz, Tim Balance, Charlotte Linnemayr-Seer, and Pavel Afanasiev. Countless other scientists who used the programs gave us constructive feedback, reported bugs or inconsistencies, and thus forced us to streamline our programs and ideas on MSA methodologies. We thank Katie Melua for inspiring the Beijing/Cambridge bicycle statistics example. Our research was financed in part by grants from: EU/NOE (grant No. NOE-PE0748), from the Dutch Ministry of Economic Affairs (Cyttron project No. BIBCR_PX0948; Cyttron II FES-0908); the BBSRC (Grant: BB/G015236/1); the Brazilian science foundations: CNPq (Grants CNPq- 152746/2012-9 and CNPq-400796/2012-0), and the InstitutoNacional de C, T&I emMateriais Complexos Funcionais (INOMAT).
Appendix
Jean-Pierre Bretaudière (1946-2008):
The Early Days of Multivariate Statistics in Electron Microscopy
Jean-Pierre Bretaudière (Figure A1) had a very fundamental influence in the early days of multivariate statistical analysis of the images of single particles in electron microscopy, an influence that was not well documented in terms of co-authorships or in other written documents. Let me explain how he was involved in the “intelligent averaging” of specific views of single particle images. Averaging of a great many “unit cells” of molecules arranged as a helical assembly [70] or as a two-dimensional crystal [71] , had at the time made quite an impression in the field and many electron microscopy groups worldwide thus started image processing projects. The first ideas of applying the concept of averaging to the images of individual macromolecules had been formulated [72] and implemented in Owen Saxton’s IMPROC software system. It was not yet clear at the time how best to find the 3D structure of a macromolecular complex based on individual two-dimensional projection images of the molecules. Would single-particle tomography such as proposed by the group of Hoppe [8] be the way towards, or did one first have to find all the different views presented by a macromolecular complex? Around 1980, we were still a decade away from answering such questions.
Since 1976, I had been employed as a project scientist in the electron microscopy group of Professor Ernst van Bruggen of the Biochemistry Department of the University of Groningen, The Netherlands. Erni van Bruggen had been most impressed by the early “averaging” successes of the group around Aaron Klug in Cambridge and wanted to incorporate those techniques in his research. It was my task to develop image processing software for data collected either with the transmission electron microscope (TEM) or the Scanning version of that instrument (a “STEM”). In the fall of 1979, I went to visit Albany, for a 6-week period, to work with Joachim Frank on automatically finding sub-populations of images in a heterogeneous population as discussed in the main paper. We had met two years earlier at an EMBO image processing course in Basel and we had been corresponding ever since. We both agreed that when averaging images of individual macromolecules, the presence of a mixture of different views was a serious and important complication: a challenging problem in need of a solution. I travelled to Albany taking with me two different sets of images in which different views could already be distinguished visually. One set consisted of images of worm hemoglobin (two different preparations: one with a clearly visible extra subunit in the middle and one without) and the other dataset of arthropod 4 × 6 hemocyanins where the difference between “flip” and “flop” could be directly seen (Figure A2).
What followed were many nights in the windowless catacombs of the New York State Department of Health (Division Laboratories and Research, now the Wadsworth Centre) beneath the Empire State Plaza. During these late sessions I met Jean-Pierre Bretaudière (“JP”) while working at the same PDP-11 45 computer (Figure A3 and Figure A4). We first talked about other issues than science, such as his personal ties to Paris (Figure A1)
![]()
Figure A1. Jean-Pierre Bretaudière in front of a Paris poster. Photo: Jan Galligan, Albany 1979.
![]()
Figure A2. Wadsworth Computer Centre, 1979. Visible on the image monitor (controlled by the 24 × 80 characters VDU) is a gallery of 4 × 6 arthropod hemocyanin images used for the first correspondence analysis of EM images. Already in this gallery one manages to directly see the difference between the “flip” and “flop” versions of the 4 × 6 half molecules of the Limulus polyphemus hemocyanin. Photo: MvH, Albany 1979.
![]()
Figure A3. Wadsworth Computer Centre, 1979. PDP-11/45 “minicomputer”. Photo: MvH, 1979.
where I had spent most of 1971 as a student. We also discussed our actual work and I explained the first attempts Joachim and I were making at separating subsets of images from the overall population of molecular images. Jean-Pierre explained what he was doing at the State Department of Health: quality control of medical laboratories in the state of New York. The Health Department would send out identical blood samples to many different medical laboratories. The results of the various chemical tests from the different labs were then compared using a French multivariate statistical technique called “l’analyse des correspondances” [73] . At the time I had never heard of multivariate statistical analysis (“MSA”), let alone of Benzécri’s “correspondence analysis” [74] .
Jean-Pierre was immediately convinced that his “AFC” program (for: “Analyse Factorielle des Correspondances”) would be able to help solve our problem and handle our “massive” amount of image data. The AFC program was a modified version of the correspondence analysis program “CORAN” by Jean Paul Benzécri [74] .
![]()
Figure A4. Jean-Pierre Bretaudière, Wadsworth Computer Centre, 1981. Photo: Robert Rej.
What was “massive” in this context was a very relative and very dated concept: at the time we had less than 64 images of 64 × 64 pixels = 4096 pixels each; that is a total data set size of less than one megabyte (each image being 16 Kbyte in floating point format). An uncompressed picture from even the cheapest digital camera today is much larger than one megabyte! However, it was characteristic for those early days in computing, that the 16-bit PDP-11 computer would only address 216 bytes = 64 Kbytes. It was thus difficult to fit any “major” computational problem in the central memory of such a computer! The critical memory requirement in our case was the “core” needed for the square variance-covariance matrix (see main article) which, for 64 images would already occupy 16 Kbyte of the precious available address space.
As we sat together with Joachim the following morning to discuss the matter, we were all convinced that this was very much worth trying. Joachim wrote a small conversion program to allow JP’s AFC program to read the aligned images produced by the SPIDER program. The result of this first correspondence analysis of single particles was an instant success!
The first trial was on a giant annelid hemoglobin data set extracted from images of hemoglobins from two different species. The giant annelid hemoglobin of Oenone fulgida visibly had extra density in the centre [75] compared to the well known giant hemoglobin of the common earthworm Lumbricus terrestris which rather had a “hole” in the centre of its double-ring geometry. The separation of the two species, artificially mixed for this analysis, was so obvious that we immediately moved to a single data set with real internal variations. We thus moved to the second data set I had brought from Groningen, negative-stain images of Limulus polyphemus 4x6-meric hemocyanins (a dissociation product of the full 8x6-meric native hemocyanin).
On the correspondence analysis maps of these images four clear groups (“classes”) were visible. These reflected the “flip and flop” versions of the 4 × 6 oligomer where the right hand 2 × 6 part can be shifted up or down with respect to the left-hand side 2 × 6-mer. This behaviour had already been anticipated based on visual inspection of the images (Figure A2). However, that effect would have led to a subdivision into two groups rather than four. The other effect, which I had not anticipated, split both the “flip” and the “flop” groups in two, namely that of “rocking”. The four hexamers constituting the 4 × 6-mer were apparently not in one plane which led to two stable “rocking” positions where 3 out of 4 hexamers would be in direct contact with the carbon support film. This explanation, however, was not so straightforward because the alignment algorithm used at that time would itself introduce a 180˚ ambiguity in the in-plane rotational alignment. Both the shape of the molecule and the flaws of the alignment scheme thus led to the same effect. This issue was eventually resolved after confusing argumentations with referees [11] [12] . The books that JP recommended [74] ; and especially [14] became my “bibles” for years to come (English versions of these books have since appeared making that literature more accessible).
In these early MSA results, we would print out two-dimensional maps of the positions of the images on the first factorial co-ordinates and then draw a circle around a cluster of images, call that a “class” and then sum the members of that class for further interpretation. This visual classification, after eigenvector-eigenvalue data reduction, obviously had its shortcomings and was impossible to perform when more than ~3 factorial axes were to be taken into account. It was JP who also pointed me in the direction of automatic classification and the wealth of literature on the subject [76] , which then led to the introduction of automatic Hierarchical Ascendant Classification, “HCA”, in electron microscopy [16] .
Jean-Pierre was a wild guy-bursting with energy, full of ideas and projects, always burning the candle at both ends. I have vivid memories of, after a late evening session at work and an even longer night in a bar, stumbling out of the bar roaring with laughter along with JP and Vicky, Joachim’s programmer who later became JP’s wife. JP was always exuberant in his love for life and all its adventures. Jean-Pierre, having seen the success of correspondence analysis in single-particle electron microscopy, became directly interested in EM image processing. He changed fields and moved to the University of Texas Medical Center in Houston, Texas, where he developed the SUPRIM image processing system for electron microscopy [77] . In Houston, being JP, he was proud to declare himself more Texan than the Texans: he bought a ten-gallon hat, a huge Lincoln with “BRETO” vanity plates, and a large gun to defend his home and family. I still vividly remember the self-satisfied smile on his face when he told me this...
The beautiful portrait of JP by Jan Galligan (Figure A1), which so clearly documents his joie de vivre, resurfaced in my archives a few years ago. After scanning it, I searched the web to find JP’s current email address hoping to put a smile on his face. Unfortunately, I failed to find a current address. Only recently, after receiving the sad message of Jean-Pierre’s death, did I learn he had moved to Madagascar, after selling his very successful French computer business-Brett Computers-moving away from western society’s rat-race. Some say that scientists are dry and boring people… There is no doubt that with JP no longer around, the world of science has become a slightly more boring small universe.
I am grateful to Jan Galligan and Robert Rej for letting me to use their pictures, and for their support and constructive suggestions (Marin van Heel; 2009).
![]()
Submit or recommend next manuscript to SCIRP and we will provide best service for you:
Accepting pre-submission inquiries through Email, Facebook, LinkedIn, Twitter, etc.
A wide selection of journals (inclusive of 9 subjects, more than 200 journals)
Providing 24-hour high-quality service
User-friendly online submission system
Fair and swift peer-review system
Efficient typesetting and proofreading procedure
Display of the result of downloads and visits, as well as the number of cited articles
Maximum dissemination of your research work
Submit your manuscript at: http://papersubmission.scirp.org/
NOTES
![]()
*A preliminary version of this publication was distributed as part of the final report of the EU-funded 3D-EM Network of Excellence on a DVD called: “Electron microscopy in Life Science”, A Verkley and EV Orlova (2009). This paper is dedicated to the memory of Jean-Pierre Bretaudière (see Appendix).