
1. 引言
人类癌症的产生原因往往和基因的变化密不可分,而单个或是几个基因的变化不一定会导致癌症的产生。癌症的产生机制十分复杂 [1] [2],与生物系统层面的变化相关。本文目的就是从系统层面研究与癌症相关的关键基因。因此,我们构建了分子网络也就是文中的蛋白质互作网络来反应系统层面癌症的特性。SSN方法 [3] 是本文用来构建蛋白质互作网络的方法,它有着传统网络方法不具备的优点。传统网络方法主要是利用基因测序结点的表达数据来筛选差异基因、构建网络的,这样做也确实能反应出一些和癌症有关的关键基因,但是由于此方法只将样本分为总的两类进行比对,即正常样本和癌症样本,因此无法关注每一个癌症样本的表达模式。然而,SSN方法使用正常样本作为对照,可以构建每个癌症样本的蛋白质互作网络。这么做的好处是我们可以具体关注每个癌症样本的癌症表达模式,以此实现针对每个癌症个体的个性化医疗。
高通量测序技术,也称下一代测序技术 [4],有着测序准确度高、测序效率高以及测序成本低等优势。测序技术的发展,使得获取大量的基因表达数据成为了现实。本文主要的研究数据就是基因表达数据,我们从TCGA数据库 [5] 下载了8种人类癌症的高通量测序 [4] 数据用以SSN的构建。在构建SSN时,我们先用Matlab进行数据分析和预处理,之后使用了String网页工具来获得蛋白质互作网络图 [6]。构建出SSN后,我们使用Cytoscape软件 [7] 提取各癌症样本的TP53子网。TP53子网是我们重点关注的对象之一,这是由于TP53基因是为制造p53这种蛋白质提供指令的基因,而p53蛋白质的作用就是抑制肿瘤,也就是说TP53基因与癌症的产生是息息相关的 [8] [9]。后续我们对比了SSN方法和传统网络方法在分类正常样本和癌症样本时的情况,其中SSN方法通过Matlab编程实现差异基因的筛选,而传统网络方法通过NetworkAnalyst网页工具 [10] 实现差异基因的筛选。筛选完差异基因后我们提取了前五位的差异基因,通过Heatmapper网页工具 [11] 进行热图的制作以及分类准确率的统计。
我们使用SSN方法完成对癌症表达模式的分析,是对传统网络方法的一种改进与提升,并且我们也验证了SSN方法的可用性和可靠性。
2. SSN
2.1. SSN简介
对于人类而言,大多数疾病的产生机制、产生原因都是复杂的,尤其是对于发病原因仍未明了的癌症来说。这些复杂的疾病不单单由一个分子或者几个分子的功能失常引起,而是由于系统的失常或者分子网络的失常导致的。因此,如果想要对疾病的产生机制进行研究,我们主要的研究对象应当是系统或者分子网络。因此,本文的主要研究对象就是分子网络,我们利用了分子网络来反应系统层面的癌症的表达模式。分子网络是一种分子之间相互作用的关系网络,它用来反应分子之间相互连接的关系。分子网络包括了生物代谢与信号传导网络、基因转录调控网络以及蛋白质互作网络,其中本文使用的分子网络是蛋白质互作网络。蛋白质互作网络是由单独的蛋白通过彼此之间的相互联系、表达作用形成的网络,它能反映不同基因表达的蛋白质之间联系的强弱,我们可以用它来反映系统层面疾病的性质。图1是大肠杆菌有机体系中四种蛋白质相互联系形成蛋白质互作网络的例子。每个结点trpA、trpB、trpC、trpD均表示一种蛋白质,结点之间的连线(边)代表蛋白质之间的联系关系。此外,连接两个结点的边还能用来反应这两种蛋白质联系的强弱性质,边越粗说明它们之间的联系越强。
![]()
图1. 蛋白质互作网络示例。
为了构造癌症的蛋白质互作网络,我们需要大量的基因表达信息,这完全得益于高通量测序技术。高通量测序技术,即第二代测序技术,是一种对基因测序、获取基因表达信息的测序方法。第二代测序技术对比第一代测序技术,有着可靠性更高、测序成本低廉、一次能测定几百万条DNA分子等优点。也正是由于测序技术近些年的飞速发展,人们越来越热衷于研究分子网络,尤其是对于疾病的分子网络的研究。并且用分子网络来阐明疾病系统层面的一些特质也正受着越来越多的关注。因此,本文的最终目的是利用不同癌症的基因测序信息构造癌症样本的分子网络,然后通过研究这些分子网络,来寻找不同种类的癌症是否具有不同的、独特的网络模式,并且尝试通过分子网络去寻找癌症发病的一些关键因素。
SSN就是本文用来研究癌症疾病模式所构造的蛋白质互作网络。SSN是基于一组正常样本对照所构建出来的蛋白质互作网络。高通量测序数据中,存在癌症样本和正常样本,一个正常样本的数据代表从一个正常人身上提取mRNA进行测序获得的数据,一个癌症样本的数据代表从一个患癌的人身上提取mRNA进行测序获得的数据,样本的数量代表的就是人的数量。为了构建SSN,我们先利用一定数量的正常人的样本作为对照,通过基因之间的相关性,我们以这些作为对照的正常样本为基础构建一个网络,我们将这个网络命名为参考网络。这个参考网络只是构建SSN中用来过渡的网络,因此不需要将它具体化(用结点和边的形式画出来)。为了获得正常样本中两个基因之间的相关性,我们通过计算两个基因之间的皮尔逊相关系数,以此来量化相关性。只要得到所有基因的两两对应的相关性,那么显然我们就能构建这些基因表达的蛋白质所对应的蛋白质互作网络,即参考网络。接着我们引入了一个癌症样本,再计算正常样本加入癌症样本后的两个基因的相关系数,如果相关系数变化很大,那么我们可以初步认为这两基因的变化与这个样本癌症的产生有关,这是因为正常样本有共同的基因表达模式,即使有差别相关性也不会变化很多。我们将以正常样本加上一个癌症样本为基础构成的网络称为扰动网络。同样这个扰动网络也只是构建SSN中用来过渡的网络,因此也不需要将它具体化(用结点和边的形式画出来)。如上过程重复对每个癌症样本进行处理,如果大部分的癌症样本里的这两个基因都有这种相关性的变化,那么不难推断这两个基因很有可能是导致正常样本变成癌症样本的基因。通过筛选这一部分基因,我们就能获得与癌症产生相关的关键基因。
这个方法里,正常样本作为对照使用,实际上我们构建网络使用的样本就仅仅只有一个癌症样本。与传统构建网络的方法对比之下,传统构建网络的方法往往需要大量的样本。并且在筛选差异基因部分,传统网络方法使用的是对结点进行筛选,SSN方法使用的是对边进行筛选,据资料显示,SSN方法能筛选出一些传统网络方法不能筛选出的关键基因,这也是SSN方法相对于传统网络方法的优点。传统网络方法是将正常样本和癌症样本全部分类放在一个矩阵,比如1到20列是正常样本的表达数据,21到40列是癌症样本的表达数据。每一行代表一个基因测序结点,通过逐行计算正常样本和癌症样本之间值的倍数FoldChance简记为FC,和检验p值以及通过一些方法修正的p值,然后通过框定条件:
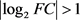 (2-1)
(2-1)
(2-2)
筛选出关键的差异基因,以此来构建蛋白互作网络。相对于传统网络方法,SSN方法最大的特点还是在筛选差异基因的时候只使用了一个癌症样本。由于SSN方法仅仅使用一个癌症样本来研究癌症的疾病模式,并且对于每种癌症的每个癌症样本均能生成一个对应的蛋白质互作网络,所以我们将这个用扰动网络减去参考网络得到的差异网络命名为单样本网络(Single Sample Network),这也是SSN这个名字的由来。这里差异网络的意思就是扰动网络减去参考网络得到网络的名称,因为由两个网络表达量之差得来,所以称之为差异网络。如果我们对每个癌症样本都去计算基因之间的两两相关性,意味着实际上我们可以对每个癌症样本构建差异网络。这是SSN方法相对于传统网络方法的优势之处,这也是我们使用SSN方法的很重要的原因。由SSN方法我们可以得到每个样本自己独特的、特异性的表达模式,因此我们也将这个差异网络称为样本特异性网络(Sample Specific Network)。
综上所述,我们使用SSN方法的目的,就是建立一个通过一系列数据处理得到的分子网络来对人类癌症疾病模式进行研究,以此实现对传统网络方法的一种补充、改进,弥补传统网络方法在样本需求高、筛选差异基因构建蛋白互作网络等方面的一些缺陷。
2.2. 背景知识
2.2.1. 皮尔逊相关系数PCCs
构建SSN时,为了将两个基因之间的相关性量化,我们引入皮尔逊相关系数来计算,通过数值定量反应基因之间的相关性。
皮尔逊相关系数是用来计算两个变量之间线性相关性的一个值,它的定义是两个变量之间的协方差与标准差的商。
(2-3)
假设有两个变量X、Y,如果X、Y正相关,那么它们的皮尔逊相关系数等于1;如果X、Y负相关,那么它们的皮尔逊相关系数等于−1;如果皮尔逊相关系数等于0,那么X、Y不相关。本文主要使用皮尔逊相关系数的绝对值,如果皮尔逊相关系数的绝对值向0靠拢,那么两个变量的相关性减小;如果皮尔逊相关系数的绝对值向1靠拢,那么两个变量的相关性增加。
本文中参考网络由n个正常样本构成,那么我们定义基于这n个样本的两个基因的相关系数为
,于是在参考网络中连接这两个基因的边对应的值就为
;而扰动网络是由n个正常样本以及1个癌症样本构成,那么我们定义基于这n + 1个样本的两个基因的相关系数为
,于是在参考网络中连接这两个基因的边对应的值就为
。差异网络是扰动网络减去参考网络而来的网络,因此在差异网络中连接这两个基因的边的对应值就为
,记为
。
2.2.2. t检验
在说明
分布情况时,可以用自由度为n − 2的检验来计算
的p值,检验统计量为
(2-4)
其中p值的意思是落在拒绝域的概率。本文作出了当
时
的分布情况,即概率密度函数,如图2所示。当n增加的时候,
的分布会越来越趋近正态分布,于是我们猜测,当n趋向于无穷的时候,
的分布无限接近于正态分布。
2.2.3. u检验
本文中使用u检验来检验SSN中的边是否显著,如果检验p值小于0.05,那么意味着落在拒绝域的概率很小,则认为这条边在SSN中显著;反之如果检验p值大于0.05,则认为这条边在SSN中不显著。一条边在SSN中显著也就意味着这条边在SSN中是存在的。本文中一些地方,比如绘制人类8种不同癌症的各10个随机癌症样本的TP53子网,筛选差异基因时,由于筛选出的差异基因数量超出了String网页工具能处理的数量,我们将P值范围区分为小于0.01和大于0.01,小于0.01则边在SSN中显著,大于0.01则边在SSN中不显著。
![]()
图2.
时
的概率密度函数。
因为
的分布在n比较大的时候趋向于正态分布,那么显然
也是趋向于正态分布的。
(2-5)
我们通过检验
与
的总体均值0是否相等来判断一条边是否显著,实际上我们判断的是
到
的变化是否显著。因此我们实际上进行的是两个正态总体值的假设检验,即检验的H0为
,H1为
.那么我们可以将其转变为H0是
,H1是
,于是这就变成了单个正态总体的假设检验,
就是
,于是假设检验就变为验
是否等于0。
单个正态总体的假设检验中,u检验的检验统计量为
(2-6)
本文使用的u检验为双边u检验,如图3所示,正态分布的双尾区域(打阴影部分)即为拒绝域,落在这两部分的概率即为p值。我们知道正态分布的概率密度函数总面积为1,即整个图的面积为1,那么p值就等于落在双尾区域阴影部分的面积的值。
2.3. 构建单个癌症样本的SSN
SSN,代表单样本网络(Single Sample Network),也代表样本特异性网络(Sample Specific Network)。由于癌症产生的原因是复杂的、系统性的,我们需要构建不同种类癌症的分子网络,观察不同的癌症是否有不同的网络模式,并且通过构建的分子网络来寻找与发病有关的的基因或是蛋白质。SSN就是本文我们构建的用来研究癌症疾病模式的分子网络,也就是蛋白质互作网络。
为了构建一个癌症样本的SSN,我们首先需要构建一个基于普通人的用以
![]()
图3. 正态分布的概率密度函数。
对照的网络,即参考网络。参考网络是由一组正常的样本为基础构建得来的,正常样本的选取数量在10~20个左右,由于一些癌症的正常样本数不多,于是选取的正常样本数量极个别会有例外。参考网络的正常样本数如果太多,那么计算的速度就会减慢;数量如果太少,那么在计算基因之间的相关系数以及判断相关系数是否显著变化的时候,精确度就会降低。由于参考网络中所有的样本都是正常样本,因此参考网络是具有正常样本公共属性的网络。我们构建参考网络的目的是将参考网络作为扰动网络的对照。扰动网络指的是通过在正常样本种加入一个癌症样本,以这些样本为基础构建的网络。我们将扰动网络去和参考网络作比较,观察在两个网络中连接两个相同基因的边是否有显著的变化。如果有显著的变化,那么我们初步认为这个变化和癌症的产生是有关的;如果没有显著的变化,就认为与癌症的产生无关。对照的网络需要是正常的、普遍的,因此构成参考网络的样本中不可以有癌症样本。
我们从TCGA数据库下载下来TCGA数据后,经过Excel简单处理的结果一般如图4中样本123构成的表所示所示,第一行代表的是样本的名称,这里用s1、s2……来表示,第一列代表的是基因测序节点,这里用g1、g2……来表示。因为样本中正常和癌症样本是穿插出现的,所以我们要先对正常样本以及癌症样本分类,分类详细过程见数据预处理部分。分类完正常样本与癌症样本后,我们就可以选择一定数量的正常样本通过皮尔逊相关系数来构建参考网络。当一个癌症样本(图4中的d样本)被加入参考网络的时候,我们称这个加入后由皮尔逊相关系数构成的网络为扰动网络。由此,我们可以得到参考网络以及扰动网络之间的差异网络,并且参考网络与扰动网络之间的差异仅仅在于癌症样本d。
统计学中,皮尔逊相关系数PCC被用来度量两个变量X、Y之间的相关性,参考网络中两个基因(比如g1与g2)之间的相关性就由皮尔逊相关系数决定,记为
(n个样本的皮尔逊相关系数)。同理扰动网络两个基因之间的相关性就由皮尔逊相关系数决定,记为
(n + 1个样本的皮尔逊相关系数)。如果加入的样本d在基因表达的形式上与正常样本差距不大,那么
和
之间不会有显著的差异;如果加入的样本d在表达形式上与正常样本差距显著,那么
和
之间会有显著的差异。因此我们主要关注的是
和
之间的差异,即
(
),以此来构建一个差异网络。这个差异网络就是仅使用了一个癌症样本d构建出来的SSN,我们称它为样本d的SSN。此外,这个SSN只由样本d构成,所以它能单独反应样本d的癌症的疾病状况,这一点是传统网络方法不具备的性质。构建单个癌症样本SSN的算法流程图如图5所示。
2.4. 理论基础
2.4.1. PCC的分布情况
要说明SSN的理论基础,我们首先要了解PCC的分布情况。我们假设有n个样本,两组基因表达X与Y。两组基因表达分别为
,
,其中
代表基因X在第i个样本中的表达量,
代表基因Y在第i个样本中的表达量。根据皮尔逊相关系数的定义,这里
![]()
图4. 构建一个癌症样本d的SSN。
(2-7)
其中
(2-8)
(2-9)
那么可以得到
的期望为0,方差为
。因此,
的p值可以通过自由度为n − 2的t检验来计算得到,假设检验统计量为
(2-10)
2.4.2. ΔPCCn的分布情况
由于SSN是差异网络,主要研究的是
的情况,所以我们要进一步说明
![]()
图5. 构建癌症样本d的SSN的流程图。
的分布情况。我们假设有n个样本,两组基因表达X与Y。两组基因表达分别为
,
,其中
代表基因X在第i个样本中的表达量,
代表基因Y在第i个样本中的表达量。根据皮尔逊相关系数的定义,这里
(2-11)
其中
(2-12)
(2-13)
当加入一个样本d后,我们假设
,
,其中
代表基因X在第i个样本中的表达量,
代表基因Y在第i个样本中的表达量。根据皮尔逊相关系数的定义,这里
(2-14)
其中
(2-15)
(2-16)
通过线性化等一系列处理可以得到
的期望为0,标准差为
。
因此,根据中心极限定理,即统计学中用来分析随机变量序列的分布服从正态分布的定理,我们可以使用u检验(即z检验)来估计
的显著性。通过程序展现、比较
的分布与正态分布,我们可以看出分布的双尾区域是相似的。图6(a)展示了
时
的分布(红线)和正态分布(蓝线)的概率密度函数,当n增加时,
的分布与正态分布越来越接近,如图6(b)。于是我们认为当n趋向于无穷的时候,对于双尾区域来说,
的分布无限接近于正态分布。我们将图中红线的分布命名为“火山分布”。由于在做假设检验的时候,我们用的是双边u检验,因此当双尾区域相似时,我们可以简单地将这种“火山分布”当作是正态分布分析,这是我们重点关注双尾区域的原因。
因为我们要研究每条边的显著性,所以我们对每个
都要进行假设检验。原假设为
等于
的总体均值,这里
的总体均值取0。假设检验统计量为
(2-17)
如果假设检验得到的p值小于0.05,那么我们认为
所对应的这条边是显著的,即这条边在我们构建的SSN中是存在的;否则我们认为这条边不显著且不存在于我们构建的SSN中。
为了验证我们得到的u检验统计量,我们随机生成两组数当作表达数据
,
。长度n由5变化到200,X、Y之间的皮尔逊相关系数PCC从0变化到0.9。对于每一对n和PCC,都随机模拟20000次,每次模拟都选取“火山分布”双尾区域中p值等于0.05时的
的值,并且根据正态分布理论计算,我们也能得到
相对应的理论值。通过比较随机模拟的
的值和理论
的值,如图6(c),我们发现在n比较
![]() (a) n = 100时的分布情况
(a) n = 100时的分布情况 ![]() (b) n = 1000时的分布情况
(b) n = 1000时的分布情况 ![]() (c) p = 0.05时ΔPCCn随的变化情况
(c) p = 0.05时ΔPCCn随的变化情况
图6. (a)展示的是n = 100时ΔPCCn的分布(红线)和正态分布(蓝线)的概率密度函数,(b)展示的是n = 1000时ΔPCCn的分布(红线)和正态分布(蓝线)的概率密度函数。(c)展示的是当p值取0.05时ΔPCCn随着n增加而改变的曲线,其中横坐标代表n,纵坐标代表在对应的n的值以及p值等于0.05的时候所对应的ΔPCCn的值,红色曲线是理论上的曲线,蓝点曲线代表随机模拟得到的曲线。
大的时候,随机模拟曲线和理论曲线相差不大。因此u统计量的合理性得以验证,于是SSN方法理论上的合理性也得以验证。
3. 数据获取和预处理
3.1. 背景知识
3.1.1. String网页工具
在String网页工具(https://string-db.org/)中导入一列蛋白质的名称就能导出它们之间相互作用的关系以及作用的强弱,以图的结果呈现,也可以导出成tsv等格式放入Cytoscape软件里操作。
3.1.2. Cytoscape软件
Cytoscape软件是本文在用于研究不同癌症的TP53子网表达模式中使用的软件。具体使用过程是在使用String网页工具获得tsv文件后,我们将tsv文件导入Cytoscape,然后对网络进行选取我们想要的部分、更改结点和边的样式、给相关获得边和相关缺失边分别上色。以此做出少量癌症样本的清晰明了的TP53子网。此外xlsx等格式的文件也能导入到Cytoscape软件中进行作图。
3.1.3. API应用程序接口
API是Application Programming Interface的缩写,翻译过来就是应用程序接口,它是一些预先定义好的函数或是软件系统不同组成部分衔接的约定。API有如下优点,第一它能完成利用程序调用网站的数据或者处理数据,第二它能使人更加详细地了解网站内部工作机制的一些细节。如果需要画出每个癌症样本的SSN,那么就要调用String网页工具的API,因为以BRCA (乳腺浸润癌)为例,我们有1100个癌症样本,如果通过手动在网页工具上完成,需要很长时间,并且完成的都是无意义的过程重复的工作。而调用API,我们只需先将每个癌症筛选出的差异基因导出到Excel表格中,做一个循环就能使电脑自动完成这些工作。此外在绘制8种不同癌症的各10个随机癌症样本的TP53子网部分,以BRCA为例,我们程序的设定是编写了了重复次数为10次的循环,每一次循环调用一次String网页工具的API导出一个癌症样本的蛋白质连接的数据,那么一次运行就能完成一种癌症10个癌症样本的数据获取。总共80个癌症样本的数据获取也能很快解决,这就是API给我们带来的方便、快捷性。
3.2. 数据获取和预处理过程
TCGA数据可以从GDC官网(https://portal.gdc.cancer.gov/)获取数据,但是GDC官网下载需要配合下载工具使用并且操作起来比较麻烦,因此本文从Firehose (http://gdac.broadinstitute.org/runs/stddata__2016_01_28/)直接下载了8种癌症的RNA测序数据。需要注意的是Firehose现在的数据库已经不再更新,所以直接从官网选择Dashboards的Standard Data选项是无法进入网站获取数据的。由于基因测序数据种类很多,为了简便,我们选择的均为Illumina公司的RNASeqV2测序数据。从数据库中下载下来的是gz格式的压缩包文件,我们可以解压后用txt打开复制到Excel里创建csv文件,也可以用R语言对.gz格式的压缩包进行处理、提取矩阵、导出到Excel。8种癌症的名称以及测序数据的总样本、癌症样本、正常样本数以及参考样本数如表1所示。
获得样本测序数据后,我们接着要对数据进行第一步的处理。由于样本中正常和癌症样本是穿插出现的,所以第一步是对正常样本以及癌症样本分类。
TCGA样本名称如图7所示,我们只需关注第四个字段(图中01C)中的数字,这个数字范围是01~29,如果在01~09就是癌症样本,在10~29的就是正常样本。
TCGA数据下载下来后,我们发现基因测序节点名称为比如:TP53|7157,实际我们使用的只有TP53,因此需要用Matlab对每个名称进行简单地处理,思路是将|以及后面的部分全部删除即可。
![]()
表 1. TCGA数据库使用的数据
![]()
图7. TCGA数据库下载下来数据样本的命名格式,我们需要了解这个格式才能对样本进行分类。以图中代码为例,第一个字段TCGA代表的是项目名称;第二个字段02代表组织来源代码;第三个字段0001代表参与人员;第四个字段01代表样本号,C代表样本序列种样本阶数;第五个字段01代表部分的次序,D代表被分析物;第六个字段代表板的次序;第七个字段代表测序中心。
筛选差异基因之后我们要生成蛋白质互作网络,这依托的是String网页工具。这个网页工具能将导入的基因名称,通过它数据库的连接关系,来获得我们需要的基因之间的连接关系。因为我们通过皮尔逊相关系数构建的边很多在蛋白质互作图中实际是不存在的,所以我们需要借助String这个工具获得真实有的边,剔除实际上不存在的蛋白质之间连接的边。具体操作是首先选择Multiple Protein,然后导入需要的蛋白质名称清单,之后由于研究的是人类癌症,Organism选择的就应该是Homo Sapiens选项。获得图后进行confidence值的筛选以及隐藏没有连接的基因,之后就能导出我们需要的表格数据,最后利用Cytoscape软件就能美化蛋白质互作图或者提取我们想要研究的一部分。
然而,上述利用String网页工具的方法对于我们后续数据比较多的研究来说不太适合,因为在网页上直接做图每次就只能做一张蛋白质互作图,之后筛选confidence大于0.9的边以及去掉没有连接的结点,最后导出tsv格式,再将tsv格式导入Cytoscape软件,这都需要鼠标点击完成,这是一项非常耗费时间的工作,也就使得我们没有办法完成批量处理,没办法画出我们需要的每个癌症样本的蛋白质互作图。因此,通过对String网页工具帮助文件的查看与学习,我们发现可以调用网站的API来用Python程序完成上述一系列需要鼠标点击完成的工作。如图8就是我们调用String网站API完成的
![]()
图8. 通过筛选差异基因后,用Python调用String网页API接口完成的BRCA_A0CV癌症样本的蛋白质互作图。其中我们只保留了“highest confidence”的边(结点之间连接值大于0.9的边),并且隐藏了没有边连接的结点。
BRCA_A0CV癌症样本的蛋白质互作图。
4. SSN可以反映出单种癌症特有的网络形式
4.1. 前提条件
我们从Firehose下载了8种不同类型的癌症的数据。对于每种类型的癌症,我们将正常样本和癌症样本进行分类,之后取一定数量的正常样本作为参考样本来构建参考网络。接着就能通过参考样本构建每个癌症样本的扰动网络,以此就可以构建每个癌症样本的SSN。为了区别癌症的网络形式,我们首先对SSN网络中的边进行一个定义、分类。这里要注意的是,只有显著的边才能被初步认为是SSN网络中的边。一条边显著的意思就是在用u检验检验这条边对应的
与0是否相等的时候,算得的p值小于0.05或是一个约定的值比如0.01,即
不等于0的边。
1、相关上调边:即相关性增加的边,从参考网络到扰动网络,
对应的边的相关性增加,意味着相关系数变得更接近0,即
的绝对值大于
的绝对值。
(4-1)
2、相关下调边:即相关性降低的边,从参考网络到扰动网络,
对应的边的相关性降低,意味着相关系数变得更远离0,即
的绝对值小于
的绝对值。
(4-2)
3、相关恒定边:相关性认为是不变的边,那么它的
,这是不显著的边,不需要花时间去研究。
4.2. TP53子网
为了研究不同癌症特有的网络形式,我们选取的是SSN中的TP53子网作为研究对象。这是由于TP53基因是为制造p53这种蛋白质提供指令的基因,而p53蛋白质的作用就是抑制肿瘤。当细胞中的DNA受到破坏时,p53在决定DNA修复还是凋亡上起着关键作用。如果DNA可以被修复,p53就会激活其他基因来修复损伤。如果DNA不能被修复,p53就会阻止细胞分裂并发出信号让细胞凋亡。通过阻止DNA突变或受损的细胞分裂,p53有助于阻止肿瘤的发展。TP53的全称是Tumor protein p53,是著名的抑癌基因,据资料显示,在超过60~70%,甚至更高比例的实体瘤患者中,TP53基因均为突变型。此外,在正常细胞中,正常的TP53基因合成的蛋白质其实是很少的,而在癌细胞中,突变后的TP53基因会合成大量的功能异常的p53蛋白质。因此,TP53子网就是我们观察癌症疾病模式的关键部分。我们在每种癌症的癌症样本中均通过随机的方法选取了10个癌症样本,接着对TP53和其它结点连接的边进行差异基因的筛选。筛选出的差异表达基因有对应的两个性质,一个是相关上调边或是相关下调边,相关恒定边由于不显著,所以直接在这步就排除掉;另一个性质是p值。我们用p值从小到大排序这些筛选出的基因,如果数量大于1500个,我们就只取前1500个基因;如果小于那么就取全部个数,这是因为String网页工具对检索的蛋白质数量有一定的要求,而且数量越多程序就越慢。10个癌症样本筛选出的基因我们放入同一张表格的10个sheet中,例如补充文件TP53子网文件夹中的BRCA_10.xlsx文件。之后我们调用String网页工具、选取TP53子网有的边、利用Cytoscape软件将相关上调边标识为红色,相关下调边标识为绿色,最后以图片的形式导出。如图9就是我们得到的一些TP53子网的图。我们总共绘制了80张TP53子网的图,从中观察我们可以判断出,某些癌症的TP53子网几乎全是相关下调边,比如BRCA,而某些癌症的TP53子网既有相关上调边也有相关下调边,比如KIRC。在GBM癌症中,相关上调边和相关下调边数量基本对等。在LUAD、LUSC、STAD、THCA这四种癌症中,有的癌症样本相关上调边明显多于相关下调边,有的则是相关下调边明显多于相关上调边。而对于UCEC癌症,在我们随机挑选的10个癌症样本中,只有一个样本中有一条相关上调边,其余的全是相关下调边。虽然有些癌症的表达模式仅仅通过十个癌症样本无法确切地说明出来,但是我们任然能够发现,不同种类癌症的TP53子网表达模式是不同的,也就是说SSN确实可以反应一种癌症特有的表达模式。
不同种类癌症的TP53子网表达模式是不同的,SSN能反应一种癌症特有的表达形式,这个结论仅仅通过相关上调边以及相关下调边组成的形式来得出是不够的。因此我们还重点关注了TP53子网中TP53基因周围的临近基因。得益于我们筛选差异基因的代码和方法,这些临近基因具有以下两个特点:1、它们都是在某种癌症中TP53的前1500位差异基因。2、它们与TP53基因是
真实有连接关系的,也就是在String网页工具的数据库中,它们与TP53基因的combined score (同confidence)大于0.85,这也是我们筛选的条件。由于这些TP53的临近基因具有如上两个特点,我们很容易能得出一个结论,即这些基因与TP53相关性很大。而TP53基因与癌症的产生有关,因此我们不难得到:这些TP53的临近基因很可能与癌症的产生相关。接下来我们就通过分析一些TP53临近基因的性质来验证我们的观点。
我们首先关注KIRC这种癌症。KIRC是肾透明细胞癌的缩写。KIRC癌症我们也是随机挑选了10个癌症样本来绘制它们各自的TP53子网,我们使用样本0~9给这10个癌症样本命名。观察KIRC癌症中我们绘制的10个随机癌症样本的TP53子网图,我们发现除了样本1的其它9个样本的TP53子网中,TP53基因的临近基因都包含了MUC1基因。如图10所示为样本6和样本8的TP53子网,临近基因中的MUC1基因我们已用黄色标注出。查询资料我们发现,MUC1是一种比较有名的癌基因。同时我们可以观察到,MUC1基因在一部分THCA癌症样本的TP53子网中也作为TP53的临近基因存在。因此我们可以大胆地猜测:MUC1基因和KIRC以及THCA癌症的产生相关。
![]() (a) KIRC样本6
(a) KIRC样本6 ![]() (b) KIRC样本8
(b) KIRC样本8
图10. MUC1存在于KIRC癌症样本的TP53子网中。
事实上有文献验证了我们的这个猜测 [12] [13]。
在GBM癌症的10个随机癌症样本的TP53子网中,我们发现在一些样本的TP53临近基因包含了CCNB1基因、RB1基因以及CCND1基因,这三种基因均为癌基因,与癌症的产生密不可分。
在LUAD癌症的10个随机癌症样本的TP53子网中,我们发现在一些样本的TP53临近基因包含了MCL1基因、TOP2A基因、NGFR基因以及HDAC1基因,这四种基因均为癌基因,与癌症的产生密不可分。
通过我们粗略的排查,就从各个癌症的TP53子网中找到了与这些癌症产生相关的基因。并且我们挑选的一般都是比较有名的癌基因,如果一个一个基因去挑选,可以发现更多的癌基因。这有力地验证了我们的观点:TP53的临近基因很可能与癌症的产生相关。并且由于从每种癌症TP53基因的临近基因挑选出来的癌基因是不全相同的,所以我们也能得到如下结论:在SSN方法下,不同种类的癌症具有不同的、特异性的网络表达模式。
同时,对于每个癌症样本,我们都能发现它TP53子网中的有关癌基因。也就是说SSN方法使得我们能做到对每个癌症病人的发病因素进行分析,这使得我们对于不同个体能够针对性地靶向用药,这也是SSN方法很重要的优势。
4.3. 结论
SSN方法用TP53子网来反应8种癌症不同的网络表达模式。通过相关上调边和相关下调边的分类,首先我们确定了TP53子网在不同癌症下表达模式是有差异的。接着通过分析TP53子网中TP53基因的临近基因,我们发现这些临近基因很可能与癌症产生相关,并且每种癌症的TP53临近基因中包含的癌基因是不太相同的。也就意味着在SSN方法下,不同癌症确实具有不同的网络表达模式。对于每个癌症病人(每个样本),我们也能通过TP53子网判断出个体与癌症产生相关的突变的基因,做到了样本特异性,使得医疗能够做到针对个体的个性化医疗。
5. 正常样本和癌症样本的分类
5.1. 使用工具
5.1.1. NetworkAnalyst网页工具
我们使用NetworkAnalyst网页工具(https://www.networkanalyst.ca/)来实现传统网络方法对差异基因的筛选。这个网页工具的使用非常简单明了,只需按照网页给出的步骤一步一步分析,就能得到差异基因以及它们对应的FoldChange、P值等等性质的表格。使得我们不借助R语言编程就实现了传统网络方法下差异基因的筛选。
5.1.2. Heatmapper网页工具
我们使用Heatmapper网页工具(http://www.heatmapper.ca/expression/)绘制了SSN方法和传统网络方法在分类癌症样本时的热图以及样本树状图,用以比较两种方法分类的准确性。
5.2. 传统网络方法对正常样本和癌症样本的分类
传统网络方法是将正常样本和癌症样本全部分类放在一个矩阵。每一行代表一个基因测序结点,通过逐行计算正常样本和癌症样本之间值的倍数FoldChance简记为FC,一般使用的形式是它的对数形式
和检验p值,然后通过框定
和p值范围筛选出关键的差异基因:
(5-1)
(5-2)
这里我们选取了STAD癌症的220个样本,其中包含了35个正常样本和185个癌症样本。筛选出的差异基因我们根据p值从小到大排列,发现p值等于0的差异基因都有很多。接着我们对p值等于0的差异基因进行FC的从大到小排列,以此筛选出前五位的差异基因。随后我们提取只含这五个差异基因的表达矩阵,放入Heatmapper网页工具进行热图的制作。这里需要主义的是Heatmapper对表达矩阵的格式有一些要求,我们需要在表格第一行第一列输入“NAME”,最终热图格式上才不会有问题。此外,我们利用传统方法筛选差异基因用的是NetworkAnalyst网页工具。我们首先选择NetworkAnalyst网页工具中的Gene Expression Table选项,之后导入220个样本的表达矩阵,设置导入的Organism条件为Homo Sapiens,Data type条件为RNA-seq data,ID type条件为Official Gene Symbol,Gene-level summarization条件为Mean。提交数据完成后,我们点击proceed进入下一步的数据处理。简单检查下样本数量、基因数量后,进入下一步。Filtering选项我们就按默认设置,由于我们的数据从TCGA数据库下载下来的时候已经是正态化数据,因此Normalization选项选择None,提交后进入下一步。Statistical method我们选择Limma包,Primary factor选择预先定义好的CLass。这里简单介绍下定义的Class,就是在每个样本的样本名下边加一行Normal或者是Tumor的分类,这项工作在Excel里直接完成。之后选择Normal versus Tumor,提交进入下一步后,我们就能发现我们获得了一张差异基因的排序表格。由于我们只需要前5位差异基因的热图,而NetworkAnalyst网页工具之后的步骤给出的是包含所有差异基因的热图,所以我们将前五位基因名称记下以后,使用Excel表格制作只含这五位基因的样本表达数据。至此我们获得了前五位的差异基因和它们的样本表达数据,接下来我们要通过这些数据完成热图的制作。
我们借助Heatmapper网页工具来绘制热图。首先导入之前得到的220个样本前五位差异基因的表达数据,如果xlsx格式显示导入错误的话,不妨使用txt格式导入。Scale Type选择Column,Color Brightness选择50,Color Scheme选择Custom之后,我们定义基因高表达的地方为红色,低表达的地方为绿色,中间颜色为黑色。分类方法选择Average Linkage,使用欧拉距离,接着将聚类应用到行以及列上面,同时行列的树状图也全部显示。如此我们就得到了传统网络方法下分类正常样本和癌症样本的热图以及样本分类情况的树状图。
我们使用这两个网页工具的原因是,可以不用借助R语言编程来实现热图的制作,直接点击鼠标就能完成,非常方便。
导出热图以及样本分类的树状图后,我们通过数树状图中错误分类的样本,得到220个样本中有4个样本的分类不正确,以此计算出准确率约为98.2%。由传统网络方法得到的热图如图11所示。其中红色代表高表达基因,越红代表表达量越高;绿色代表低表达基因,越绿代表表达量越低。
5.3. SSN方法对正常样本和癌症样本的分类
与传统网络方法对正常样本和癌症样本分类的方法不同之处就在于筛选差异基因的方法,其它制作热图、计算准确率的过程完全相同。为了筛选差异基因,这里我们使用Matlab编程实现,具体代码详见补充文件或是附录。具体思路是35个正常样本作为对照,形成参考网络。之后依次构建185个癌症样本的SSN,如果一个癌症样本里某个基因筛选出来p值小于0.05,也就是意味着这是差异基因,那么给这个基因的计数加1。最后统计出在185个SSN里,所有基因是差异基因的次数。次数从高到低排序,以此就能获得前五位的差异基因,表2是前五位差异基因出现的次数统计。之后同传统网络方法分类的做法一样,我们将220个样本的前五位差异基因的表达数据导入到Heatmapper网页工具,以此做出热图以及样本分类的树状图。根据统计220个样本里有3个样本没有被正确分类,那么得到SSN方法分类正常样本和癌症样本的准确率约为98.6%。由SSN方法得到的热图如图12所示。其中红色代表高表达基因,越红代表表达量越高;绿色代表低表达基因,越绿代表表达量越低。
![]()
图11. 传统网络方法分类热图。
![]()
表 2. . SSN方法筛选前五差异基因。
5.4. 结论
基于STAD癌症,我们完成了传统网络方法以及SSN方法对正常样本和癌症样本的分类。从实验结果上来看,基于SSN方法的分类能够提高样本分类的准确率。
比较传统网络方法和SSN方法,我们可以看出在筛选前五位差异基因的时候,两个方法仅仅具有一种基因的不同。传统网络方法筛选出的是FAP基因,而SSN方法筛选出的是COL11A1基因。在热图方面,传统网络方法将低表达的部分聚类在了几乎最左边的位置,SSN方法将低表达部分聚类在了靠左的位置。而这些低表达的区域正是正常样本的基因所对应的位置,以此我们能看出个体如果患有STAD癌症,那么这5种差异基因很可能不是低表达的。
![]()
图12. SSN方法分类热图。
换句话说如果某个样本的这五种基因高表达了,那么这个样本很可能就是癌症样本。我们将正常样本按照NOM1-35的顺序命名,将癌症样本按照TUM1-185的顺序命名。那么在数树状图计算分类准确率的时候,我们得到SSN方法分类时,TUM20、TUM156、TUM101基因分类错误。而传统网络方法分类时,NORM25、TUM156、TUM101、TUM88基因分类错误。由此我们得到基于STAD癌症,使用SSN方法能够提高样本分类的准确率,并且准确率较高。
6. 结论与展望
完成了以上所有工作后,我们认为SSN方法在反应癌症表达模式、寻找癌症产生的关键基因以及分类正常样本和癌症样本上是可行的,并且可靠的。此外,SSN方法还具有传统网络方法不具备的优势,即SSN方法可以对每个癌症样本的关键癌症相关基因进行筛选,并且判断每个癌症样本的癌症疾病模式,以此实现医疗上能够针对不同个体进行个体化的医疗。
在筛选某一种癌症所有的差异基因时,传统网络方法总共只经过了一次总的FoldChange和p值或是修正p值的筛选,而SSN方法有几个癌症样本就需要筛选几次,再经过统计每个基因是差异基因的次数,这样做无疑比传统网络方法更准确。但是实际上目前的传统网络方法筛选差异基因是经过改进的,我们了解到R语言里有三个包能够完成差异基因的筛选,第一个是基于线性模型建模的Limma包 [14],第二个是使用经验贝叶斯估计和基于负二项模型的精确检验的EdgeR包 [15],第三个是使用类似于EdgeR的负二项模型的DESeq包 [16]。实际使用这些包时,也能准确筛选出差异基因。此外,由于对于每个癌症样本都要构建它的SSN,这相比于传统网络方法增加了很大的工作量,使得SSN方法程序的运行时间会增加很多。因此,我们得出如下结论:筛选差异基因的方法有很多,每个方法都有它的优势和弊端,只要合理使用都能发挥出这些方法的价值。
对于构建生物网络,我们认为未来会出现更多更好的办法,会不仅比现有的网络方法更准确,还能一定程度上降低工作量。研究SSN的意义不仅在于对传统方法的改进和提升以及个体医疗化的实现,还在于提出一个构建生物网络的新思路。而研究构建生物网络的意义,就在于推动对于癌症研究的发展,找出更多关键的癌基因,为人类日后研究透彻、甚至治愈癌症提供道路。
致 谢
本课题在选题及研究过程中得到程晓青老师的亲切关怀和悉心指导。她严肃的科学态度、严谨的治学精神以及精益求精的工作作风深深地感染和激励着我。从课题的选择到项目的最终完成,程老师都始终给予我细心的指导和不懈的支持,在此谨向程老师致以诚挚的谢意和崇高的敬意。