A Possible Mechanism of Cisplatin-Induced Tumor Necrosis Factor (TNF)-α Production in Murine Macrophages ()
1. Introduction
Cisplatin has been used for the treatment of various solid cancers or sarcomas; however, it can cause severe adverse effects including renal and cochlear injuries [1,2]. Nephrotoxicity, which is caused by the accumulation of cisplatin in the proximal tubule, has the potential to be a dose-limiting factor of this therapy. Previous studies reported that proximal tubular injury was caused due to increases in macrophage infiltration by cisplatin [3,4] and subsequent overproduction of the pro-inflammatory cytokine, tumor necrosis factor (TNF)-α, in tubule cells [5-7]. Thus, inhibiting the production and secretion of TNF-α in macrophages may be advantageous in reducing cisplatin-induced renal injury. The production of proinflammatory cytokines including TNF-α is mediated by the activation of mitogen-activated protein kinase (MAPK) family members [6]. MAPK pathways are cascades of serine/threonine kinases comprising three families members; extracellular signal-regulated kinase (ERK), p38 MAPK, and c-Jun N-terminal kinase (JNK), that are activated by chemical stresses and reactive oxygen species (ROS) [8-10]. The ERK signaling module is involved in cell growth, proliferation, and survival. On the other hand, the activation of p38 MAPK and JNK generally results in inflammation [8,9,11]. Ramesh et al. reported that cisplatin induced TNF-α mRNA expression via activation of the p38 MAPK pathway in renal proximal tubular cells [12]. However, the possible mechanism of cisplatin-induced TNF-α expression in macrophages has not been fully elucidated. In this study, we investigated the mechanism of cisplatin-induced TNF-α production in macrophages, with a focus on MAPK pathways, using the mouse macrophage-like cell line, RAW 264.
2. Materials and Methods
2.1. Cells Cultures
Murine macrophage-like cell line RAW 264 cells (No. RCB0535) was provided by the RIKEN RBC through the National Bio-Resource Project of the MEXT, Japan. Cells were cultured in Minimum Essential Medium Eagle (Sigma-Aldrich) supplemented with 10% (v/v) heat-incubated fetal bovine serum (Biowest, Nuaille, France), 0.1 mM non-essential amino acids (Nacalai Tesque, Kyoto, Japan), 5000 U/ml penicillin, and 5000 μg/ml streptomycin (Nacalai Tesque) in a humidified atmosphere of 5% CO2 at 37˚C.
2.2. Chemicals
Stock solutions of cis-Diammineplatinum (II) dichloride (cisplatin) and N,N’-dimethylthiourea (DMTU), which were obtained from Sigma-Aldrich (St. Louis, MO, USA), were prepared in Minimum Essential Medium Eagle (Sigma-Aldrich). Inhibitors of ERK (PD98059), p38 MAPK (SB203580), and JNK (SP600125) were purchased from Cayman Chemical (Ann Arbor, MI, USA), Jena Bioscience (Jena, Germany), and Enzo Life Sciences (Farmingdale, NY, USA), respectively. Stock solutions were prepared in dimethylsulfoxide (DMSO: SigmaAldrich) and were added to cell cultures at a concentration of 1 μM. The final volume of DMSO in each well was 0.01% (v/v).
2.3. TNF-α mRNA Assay
RAW264 cells were seeded in 12 well plates (1 × 106 cells/well) and incubated for 24 h. Cells were pretreated with the three different types of MAPK inhibitors (0.1 - 10 μM) or DMTU (100 μM) for 0.5 h, followed by stimulation with cisplatin (1 μM) for 24 h. After incubation, the expression of TNF-α mRNA was analyzed by reverse transcription polymerase chain reaction (RTPCR). Total RNA was extracted using an RNA extraction kit (Viogene, Taipei, Taiwan) according to the manufacturer’s instructions. RNA was converted into a firststrand complementary DNA (cDNA) by ReverTra Ace (TOYOBO, Osaka, Japan). cDNA was then used as a template for RT-PCR with TNF-α and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) specific primers in a thermal cycler (PC-320, ASTEC, Fukuoka, Japan). The specific primers (Invitrogen, Carlsbad, CA, USA) used in this study are listed below: TNF-α, 5’-GGG TGT GAA CCA CGA GAA AT-3’ and 5’-TTA CTC CTT GGA GGC CAT GT-3’; GAPDH,5’-GGG TGT GAA CCA CGA GAA AT-3’ and 5’-TTA CTC CTT GGA GGC CAT GT-3’. The PCR program was 30 sec, 95˚C, followed by 26 cycles of denaturation, 30 sec, 95˚C; annealing was 30 sec, 50˚C; elongation, 1 minute, 72˚C, followed by 5 minutes, 72˚C. PCR products (TNF-α: 346 bp, GAPDH: 610 bp) were separated on 1.8% agarose gels (Nacalai Tesque) and stained with a 1/10,000 dilution of SYBR Safe DNA gel stain (Invitrogen). Gels were captured with E-graph (AE-900, ATTO, Tokyo, Japan) and band densities were analyzed for quantification using the CS Analyzer ver3.0 (ATTO). The expression levels of TNF-α were normalized to corresponding GAPDH mRNA levels.
2.4. MAPK Phosphorylation Assay
Cells (6 × 104 cells/well) were pretreated with or without DMTU (100 μM) for 0.5 h, followed by stimulation with cisplatin (1 μM) for 24 h. ERK, p38 MAPK and JNK phosphorylation levels were determined using an ELISA assay kit (Cell-Based ERK1/2 (activated) ELISA Sampler Kit, RayBiotech, Norcross, GA, USA) according to the manufacturer’s instructions. The absorbance of each well was read at 450 nm using the microplate reader (Model 680, Bio-Rad, Hercules, CA, USA).
2.5. TNF-α Protein Assay
Cells were harvested in 12 well plates (1 × 105 cells/well) and treated with PD98059 (1 μM) or SB203580 (1 μM) between 0.5 h and 72 h after cisplatin (1 μM) stimulation. TNF-α protein levels in the supernatant were then determined using an ELISA assay kit (Mouse TNF-α Quantikine ELISA Kit, R&D Systems, Minneapolis, MN, USA) according to the manufacturer’s protocol. The absorbance of each well was read at 450nm using a microplate reader.
2.6. Cell Viability
Cells were cultured in varying concentrations of cisplatin between 0.01 μM and 10 μM for 24 h and cytotoxicity was evaluated by trypan blue staining (Gibco, Grand Island, NY, USA).
2.7. Statistical Analysis
Data were expressed as mean ± S.E.M. and analyzed statistically using a one-way analysis of variance followed by the Dunnett’s Multiple Comparison Test. Values of p < 0.05 were determined to be significant.
3. Results
3.1. Involvement of the MAPK Pathway on Cisplatin-Induced TNF-α mRNA Expression and Protein Production
Cisplatin dose-dependently increased TNF-α mRNA expression. A marked increase was observed with the administration of 1 μM cisplatin (Figure 1(A)). We observed no significant suppression in cell viability when cells were stimulated with cisplatin at a concentration of 1 μM (data not shown). PD98059, SB203580, or SP600125 alone did not affect TNF-α mRNA expression in RAW264 cells. PD98059 at a dose of 1 μM and 10 μM significantly inhibited cisplatin-induced TNF-α mRNA expression, but any dose of SB203580 and SP600125 did not inhibit cisplatin induced TNF-α mRNA expression (Figures 1(B)-(D)). As shown in Figure 2, cisplatin significantly increased ERK and p38 MAPK to 1.4 and 1.7-fold higher than that of controls, respectively; however, a similar response was not observed for JNK phosphorylation. Pretreatment with PD98059 and SB203580 significantly suppressed cisplatin-induced TNF-α protein production and levels in PD98059-treated cells and returned them to control levels (control: 142.6 ± 2.0 pg/ml, cisplatin: 231.7 ± 8.5 pg/ml, cisplatin + PD98059: 143.3 ± 10.9 pg/ml, cisplatin + SB203580: 177.4 ± 17.9 pg/ml) (Figure 3).
3.2. The Effect of a Radical Scavenger on TNF-α mRNA Expression and MAPK Phosphorylation
Although DMTU alone did not affect TNF-α mRNA expression, it significantly reduced cisplatin-induced TNF-α mRNA expression to the level of controls (Figure 4(A)). Furthermore, DMTU significantly decreased cisplatin-induced phosphorylation of ERK, but not that of p38 MAPK (Figure 4(B)).
4. Discussion
In this study, we demonstrated that cisplatin significantly increased TNF-α mRNA expression (Figure 1(A)). Using specific inhibitors of MAP kinase family members, we found that the transcription of TNF-α mRNA is dependent on ERK, but not p38 MAPK or JNK (Figures 1(B)-(D)). Several studies have shown that p38 MAPK and JNK are involved in inflammation and that their activation results in the production of inflammatory cytokines including TNF-α [9,13]. Our results suggest that p38 MAPK and JNK did not suppress the expression of cisplatin-induced TNF-α mRNA (Figures 1(C) and (D)). Previous reports have revealed that p38 MAPK is involved in the nucleocytoplasmic transport of TNF-α mRNA [14] and its phosphorylation causes TNF-α mRNA stabilization; thus, this activation is associated with an increase in the production of TNF-α protein.
We actually found that cisplatin induced p38 MAPK phosphorylation under the condition of increased TNF-α mRNA expression (Figure 2), and its inhibitor, SB203580, suppressed cisplatin-induced TNF-α protein production without affecting TNF-α mRNA expression (Figure 3). These findings suggested that p38 MAPK pathways are

Figure 1. The effects of MAPK inhibitors on cisplatin-induced TNF-α mRNA expression. RAW264 cells were treated with 0.01 - 10 μM cisplatin or vehicle for 24 h (A) and were treated with the ERK inhibitor; PD98059 (0.1 - 10 μM) (B), p38 MAPK inhibitor; SB203580 (0.1 - 10 μM) (C), and JNK inhibitor; SP600125 (0.1 - 10 μM) (D) between 0.5 h before and after cisplatin (1 μM) or with vehicle stimulation. TNF- α mRNA expression was analyzed by RT-PCR. Expression levels were normalized to the corresponding GAPDH mRNA levels. The mean value of the control group was set to 1.0. Data are shown as the mean ± S.E.M. (n = 3 - 6). *p < 0.05, **p < 0.01, and ***p < 0.001 significantly different from the control group. #p < 0.05 and ###p < 0.001 significantly different from the cisplatin group.
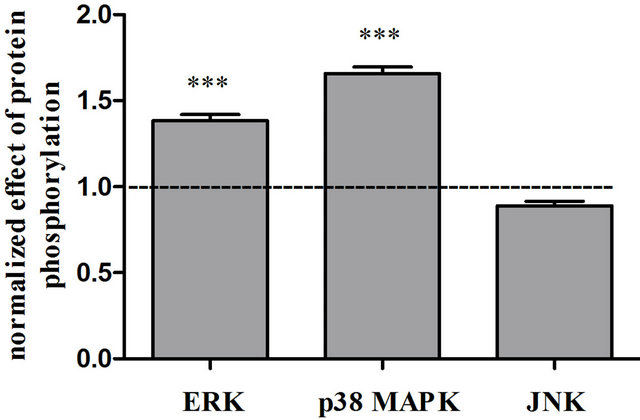
Figure 2. The effects of cisplatin on MAPK pathway activation. ERK, p38 MAPK, and JNK phosphorylation in cells treated with 1 μM cisplatin or vehicle for 24 h were analyzed by an ELISA assay kit. Phosphorylation in RAW264 cells treated with a respective inhibitor alone was defined as the value of control levels. Data are shown as the mean ± S.E.M. (n = 3 - 4). ***p < 0.001 significantly different from the control group.

Figure 3. The effects of MAPK inhibitors on cisplatin-induced TNF-α production. RAW264 cells were treated with 1 μM PD98059 and 1 μM SB203580 between 0.5 h before and 72 h after 1 μM cisplatin stimulation. TNF-α levels in the supernatant were determined using an ELISA assay kit. Data are shown as the mean ± S.E.M. (n = 3). **p < 0.01 significantly different from the control group. #p < 0.05 and ##p < 0.01 significantly different from the cisplatin group.
also closely related to the process of cisplatin-induced TNF-α protein synthesis rather than its mRNA expression.
On the other hand, although Swantek et al. reported that the JNK pathway mainly regulates the translation of TNF-α mRNA [15], we confirmed that cisplatin was not involved in the phosphorylation of JNK and SP600125 did not affect cisplatin-induced expression of TNF-α mRNA in RAW264 cells 24 h after cisplatin treatment. Sánchez-Pérez et al. reported that cisplatin induced transient activation of the JNK pathway within a few hours via the regulation of its upstream-kinase MEKK1 [16,17]. Therefore, there is a possibility that the JNK pathway may also be involved in the development of cisplatininduced renal failure at an early stage.
Rao reported that lipopolysaccharide (LPS) induced the production of inflammatory cytokines through ERK

Figure 4. The effects of a radical scavenger on cisplatin-induced TNF-α mRNA expression and MAPK pathway activation. RAW264 cells were treated with 100 μM DMTU or vehicle between 0.5 h before and 24 h after cisplatin (1 μM) or vehicle stimulation. TNF-α mRNA expression was then analyzed by RT-PCR (A) and the phosphorylation of ERK and p38 MAPK were analyzed by an ELISA assay kit (B). The mean value of the vehicle control was set to 1.0 (A) and the phosphorylation value in the cisplatin treatment group was set to 1.0 (B). Data are shown as the mean ± S.E.M. (n = 3 - 4). **p < 0.01 significantly different from the vehicle control group. #p < 0.05 significantly different from the cisplatin group (A). *p < 0.05 significantly different from the control group (B).
phosphorylation [18]. LPS phosphorylates Ras and Raf-1, upstream-kinases of ERK, and, subsequently, this phosphorylation activates ERK [19,20]. The activation of ERK regulates the expression of TNF-α mRNA via the activator protein-1 transcription factor pathway [21-24]. Shishodia reported that cisplatin enhanced the phosphorylation of Ras in macrophages [25]. From these observations, our results indicated that ERK phosphorylation is needed for cisplatin-induced TNF-α mRNA expression in macrophages.
Several studies have shown that ROS leads to increases in the phosphorylation of ERK and p38 MAPK [26-28]. Cisplatin generates hydrogen peroxide (H2O2), a relatively long-lived species of ROS, in a variety of cells including macrophages [29,30]. From these findings, we hypothesized that cisplatin-induced TNF-α production in murine macrophages may be due to an increase in ERK and p38 MAPK phosphorylation by the H2O2 generated by cisplatin. We demonstrated that DMTU inhibited the expression of TNF-α mRNA and also decreased cisplatin-induced phosphorylation of ERK. Since DMTU, an agent with a long half-life and the ability to scavenge H2O2 and hydroxyl radicals selectively, can be highly and rapidly permeable to cell membranes, it has the ability to inhibit the inflammatory response caused by ROS generated within cells [31]. Taken together, ERK activation, induced by H2O2 and hydroxyl radicals and generated by cisplatin, is essential for the expression of the TNF-α mRNA and production of the TNF-α protein.
We found that although DMTU slightly inhibited the phosphorylation of p38 MAPK, a significant inhibitory effect was not observed (Figure 4). The link between the TNF receptor (TNFR) and p38 MAPK activation has been previously reported [32,33]. Ligations of the TNFR phosphorylate MKK3/MKK6, upstream of p38 MAPK, have also been shown [34,35]. Therefore, we speculate that autocrine activation by TNF-α caused the phosphorylation of p38 MAPK.
In conclusion, our study showed that macrophages require the ERK pathway for cisplatin-induced TNF-α mRNA expression and that this pathway is phosphorylated by cisplatin-induced ROS.
5. Acknowledgements
This study was partially supported by a Grant-in-Aid for scientific research from the Ministry of Education, Science, and Culture of Japan (No. 21590279 and 23790598) and research grants from the Osaka Medical Research Foundation for Incurable Disease and the Osaka Cancer Society.
NOTES