Effect of disulfide bridges deletion on the conformation of the androctonin, polyphemusin-I, and thanatin antimicrobial peptides: molecular dynamics simulation studies ()
1. INTRODUCTION
Microbial resistance to antimicrobial tools is a serious problem nowadays since the number of antibiotic-resistant microorganisms has been increasing over the last years [1,2]. The impact caused by such challenging question has motivated various research groups to develop new drugs capable of dealing with the adaptation strategy (namely, the selection of organisms adapted to some specific medium) that these microorganisms develop along the years [3-6]. The reasons for the appearance of resistant bacterial strains lie on the excessive, and frequently inadequate, use of antibiotics [7]. However, nature itself has endowed some organisms with antimicrobial peptides, which are distinctive tools for survival [8]. In this context, the production of antimicrobial peptides is a powerful evolutionary ancient defensive weapon [9] that allows living organisms like plants, vertebrate and invertebrate animals, as well as fungi and bacteria to survive. Many organisms make use of antimicrobial peptides to defend themselves against harmful invader microorganisms. These peptides are part of the innate defense systems of the organisms [6]. An important characteristic of such peptides is their broad spectrum of action since they can be active against several kinds of bacteria, as well as against some fungi. The hemolytic effect and the potential toxicity exhibited by these peptides make their use against infectious diseases difficult. Heavy investments in research that could improve their selectivity and decrease their toxicity are necessary, so that these peptides can be made active against pathogenic microorganisms only.
The androctonin peptide is an antimicrobial agent constitutive of the cell-free hemolymph of the scorpion Androctonus australis. Its primary structure contains 25 aminoacid residues, Table 1, including eight positive charges spread all over the peptide chain and four cysteine residues that produce two disulfide bridges linking Cys4 to Cys20 and Cys10 to Cys16 [10]. Its molecular mass is 3076.7 Da. It is active against Gram-positive and Gram-negative bacteria as well as against a broad range of filamentous fungi. Androctonin concentrations up to 150 µM do not exhibit hemolytic activity in porcine and bovine erythrocytes when assayed under conventional conditions [10]. Its β-hairpin structure was determined by conventional two-dimensional nuclear magnetic resonance spectroscopy (2D-NMR) and by molecular modeling [11]. The 3D structure is deposited in the Protein Data Bank, PDB, (www.rcsb.org/pdb) under the code 1CZ6.

Table 1. Primary structures of the wild peptides and their respective mutants. The residues where the mutation was made are in bold letter.
Polyphemusin-I is an antimicrobial peptide encountered in the American horseshoe crab Limulus polyphemus. It has 18 aminoacid residues in length, Table 1, including seven positively charged residues and two disulfide bonds linking Cys4 to Cys17 and Cys8 to Cys13, as well as an amidated C-terminal arginine. It is active against Gram-positive and Gram-negative bacteria, and some fungi. Its solution structure determined using 1H-NMR spectroscopy is deposited in the PDB under the code 1RKK [12].
The thanatin peptide is an insect defense protein which has, at physiological concentrations, a broad spectrum of activity against Gram-positive and Gramnegative bacteria, and filamentous fungi. It presents a minimal inhibition concentration of approximately 1 µM for both Gram-positive and Gram-negative bacteria as well as fungi [13]. It appears in the haemolymph of the hemipteran insect Podisus maculiventris. Its primary structure contains 21 aminoacid residues, Table 1, including six positively charged residues, presenting a molecular mass of 2434.4 Da. Two cysteine residues, Cys11 to Cys18, forms one disulfide bridge. Its hemolytic activity has been tested in a conventional assay on porcine erythrocytes and it was concluded that it does not present hemolytic activity even at 40 µM [14]. Its three-dimensional (3D) structure in water was determined using two-dimensional (2D) 1H-NMR and molecular modeling methods which were deposited in the PDB under the code 8TFV [13].
In the present work, we will compare the structures of the abovementioned wild peptides with the structures of three mutants, namely andry4 (androctonin mutant), poly4 (polyphemusin-I mutant), and thany2 (thanatin mutant), whose primary structures are shown in Table 1. Our proposition is based on studies carried out with tachyplesin-I mutants, in which the four cysteine residues of the wild peptide were replaced with tyrosine residues [5,15]. In their studies with tachyplesin-I, these authors found a great similarity between the native and the mutant structures. They also reported that the deletion of all cysteines abolished the hemolytic effect while the antimicrobial activity remained unchanged. There is a great interest in the development of new antibiotic compounds, mainly ones free of cysteine residues, because these peptides are easier and cheaper to produce [5,15]. The detailed data provided in this work shall give support to a better understanding of the role of the disulfide bridges in the secondary structures of these peptides.
2. MODELS AND METHODS
The initial configurations of the three mutants (andry4, poly4, and thany2) were obtained using the PDB structures of the wild peptides and the mutations carried out with the DeepView/Swiss-PdbViewer 3.7 software [16]. The construction of the systems and the molecular dynamics simulations were carried out using the GROMACS 3.3.3 package [17] with the GROMOS96 force field [18], employed to model the peptides. The six systems were constructed by placing the center of mass (CM) of the peptides in the center of a cubic simulation box with edges equal to the peptide’s largest interatomic distance plus 4 nm. The box was fulfilled with water molecules using the SPC model [19]. Chloride and sodium ions were inserted, when necessary, into electrostatically favorable positions, in order to counterbalance the peptide charges, thus keeping the local electroneutrality of the systems. The covalent bonds of the peptides were constrained by the LINCS algorithm [20], while the SETTLE algorithm [21] was used to maintain the water SPC molecules structurally stable. The temperature (300 K) and the pressure (1 atm) of the systems (NpT ensemble) were regulated by the Berendsen’s algorithms [22], using correlation times of 0.1 ps and 0.5 ps for temperature and pressure coupling, respectively. A cut-off of 1.0 nm on the van der Waals interactions was used. The long range electrostatic interactions were calculated using the particle mesh Ewald summation method (PME) [23]. In order to eliminate bad contacts, the energy of the system was first energy minimized by the steepest descent algorithm. The simulation of each system was then carried out for 0.5 ns, under peptide atomic coordinate restrictions, with an integration time step of 0.5 fs. The next step of the simulations was carried out during an extra 0.5 ns period, still under peptide atomic coordinate restrictions, but an integration time step of 1.0 fs was employed. These two cycles of 0.5 ns each were performed in order to equilibrate the system. After that, the MD simulations were carried out without restrictions for 80 ns in the cases of androctonin and andry4; 85 ns for polyphemusin-I and poly4; 50 ns for thanatin and, finally, 70 ns for thany2, always using integration time steps of 2 fs. The Ramachandran angles in the wild peptides and their respective mutants were calculated, in order to compare their structures. The intramolecular hydrogen bonds (HB) were identified considering a donor-acceptor distance was smaller than 0.35 nm, and the angle between the hydrogen-donor and donor-acceptor vectors smaller than 30 degrees. Only HB occurrences greater than 20% of the total simulation time were considered. The radius of gyration (Rg) and the root mean square deviations (RMSD) were systematically used to analyze the structural and the evolutional behaviors of the peptides; the RMSD was obtained by least-square fitting of the peptide structures to the reference that was the initial structure of each peptide. The Rg provides global information about the packing of the molecules, while the RMSD is a measure of the similarity of a structure with its reference and also the temporal evolution of the structure. In these studies, the GROMACS analyzing tools package [24] was employed.
3. RESULTS
3.1. The Structures
The structural characteristics of androctonin/andry4, polyphemusin-I/poly4, and thanatin/thany2 were first analyzed by comparison of their Cα RMSD evolution, Figures 1, 3, and 5, respectively; and RMSD per residue, Figures 2, 4, and 6, respectively.
The RMSDs for each pair of peptides (wild peptide and its mutant) can present, or not, very distinct behaveiors, indicating that the stability of the mutant is similar, or not, to that of the wild peptide. The RMSD time-averages and Rg for androctonin, polyphemusin-I, thanatin, and their respective mutants are listed in Table 2. The pair of peptides androctonin/andry4 presents very close mean RMSD values (0.33 and 0.30 nm, respect-tively, Table 2). These results are in good agreement with the mean RMSD values per residue (Figure 2), since the profiles of the two curves are very similar. The RMSD of the androctonin Arg1 residue is larger than that of the same andry4 residue, while the Val3 residue presents a smaller RMSD in androctonin compared with andry4. All the other residues of androctonin and andry4 present very close RMSDs, as shown in Figure 2. The Rg mean values, shown in Table 2, are very close to the experimental Rg values calculated from the PDB structures. The RMSD and Rg data, together with the RMSD per residue data, are a strong indication of the structural similarities between androctonin and its mutant andry4.
The distinct behaviors of the polyphemusin-I and poly4 RMSDs, presented in Figure 3, show that the mutant seems to assume configurations closer to that of reference structure than the ones assumed by the wild
 (a) (b)
(a) (b)
Figure 1. RMSD plots for (a) androctonin and (b) andry4.

Figure 2. Androctonin and andry4 mean RMSD values per residue.
 (a) (b)
(a) (b)
Figure 3. RMSD plots for (a) polyphemusin-I and (b) poly4.
peptide. It is possible that the reference structure, which is an experimentally determined structure, is not a typical structure pertaining to a high density region of the phase space. The mean RMSD values (0.36 and 0.25 for polyphemusin-I and poly4, respectively) and their profiles show that the structure of the mutant is somehow different from that of the wild peptide. Figure 4 presents the RMSD per residue and shows that the motion of the residues in polyphemusin-I is greater compared with poly4, where they seem to be more locked. The close

Figure 4. Polyphemusin-I and poly4 mean RMSD values per residue.

Figure 5. RMSD plots for (a) thanatin and (b) thany2.

Figure 6. Thanatin and thany2 mean RMSD values per residue.
values of Rg (Table 2) together with the RMSDs for polyphemusin-I and poly4 suggest that the mutant poly4 presents structural similarities with the wild peptide polyphemusin-I, keeping the β-hairpin structure. Nevertheless, the very different profiles of the curves in Figure 4 (RMSD per residue) suggest there are no conformational similarities between them. The results of the thanatin/thany2 pair also suggest that some differences in the mutant/wild peptide structures have occurred. Moreover, some instability in the thany2 structure are easily detected in the RMSDs (Figure 5) in the 20 - 35

Table 2. Average RMSD and Rg for the wild peptides and their mutants.
ns region. The Rg values (Table 2) are very close to the experimental Rg value calculated from the structures contained in the PDB file. The RMSD and the Rg data suggest a structural similarity between thanatin and thany2, as well as the maintenance of the β-hairpin structure by the mutant. The RMSD per residue in Figure 6 shows that the profiles for thanatin and thany2 are slightly different, suggesting that they have different conformations.
3.2. The Hydrogen Bonds
The simulation results concerning the three peptidemutant pairs confirm that a peculiar pattern of crossstrand HBs contributes with their respective mutants. The HB between the residues Val3 and Arg23 that is present in androctonin, is absent in andry4 (Table 3). The lack of this HB allows increased motion of the Val3 residue in the mutant, as can be seen from the RMSD per residue (Figure 2). The same flexibility does not happen with the Arg23 significantly to the stabilization of the β-hairpin-like structure adopted by androctonin, polyphemusin-I, and thanatin in aqueous solution. This same peculiarity was observed in our previous work [25] with the simulations of gomesin and protegrin-1 in aqueous media. Tables 3, 4, and 5 summarize the results for the cross-strand HBs, together residue because, in andry4, this residue forms an HB with the Ser2 residue, as shown in Table 3. In general, HB occurrence is larger in andry4 due to the absence of disulfide bridges. These results draw attention to the significant role of the cross-strand HBs in the maintenance of the structural stability of the mutants despite the absence of the disulfide bridges. Indeed, it has been experimentally observed that the location of the disulfide bridges within the β-hairpin structure determines the β-hairpin stability only when the disulfide bonds link originally non-hydrogen-bonded residues facing each other [26]. An exception is the HB between the Arg5 and Thr21 residues, since its occurrence decreases as a consequence of the

Table 3. Androctonin and andry4 hydrogen bonds determined by molecular dynamics simulations.

Table 4. Hydrogen bonds found in the polyphemusin-I and poly4 simulations.
HB formed between the Tyr4 and Thr21 residues in andry4. Three extra HBs between the Tyr4/Thr21, Tyr4/ Tyr25, and Arg23/Ser2 residues, which are not present in androctonin, also contribute to the maintenance of the β-hairpin structure in andry4. The Tyr4 residue uses the NH and the OH (from the aromatic ring) to form HB simultaneously with Thr21 and Tyr25 residues. All the HBs found in polyphemusin-I are also present in poly4

Table 5. Hydrogen bonds found in the thanatin and thany2 simulations.
(Table 4), albeit with increased occurrences. In addition, two extra HBs are also found in poly4, between the Gly11/Tyr8 and Phe12/Tyr9 residues, promoting a relevant decrease in motion in the region of the turn (Tyr8, Tyr9, Arg10, Gly11, and Phe12), as corroborated by the RMSD per residue in Figure 4. In the absence of the disulfide bridges, the occurrence of the HB between Tyr9(NH)-Phe12(CO) increases from 76.5% in polyphemusin-I to 93.3% in poly4 (Table 4). In addition, formation of another HB between Phe12(NH)-Tyr9(CO), which is not present in polyphemusin-I, is observed, and it contributes even more to the reduction in motion, as detected by the RMSD per residue. These two new HBs play an important role in the stabilization of the folding region and are probably responsible for the differences in the observed RMSD behaviors.
Finally, in the pair thanatin/thany2, the same thanatin HBs were found in the thany2 peptide, Table 5. However, the occurrence of some HBs decreases, as in the case of the HB between the Ile8/Met21 residues, which is formed sometimes between the Ile8 NH group or alternatively with one of the CO groups of Met21. These interactions are not simultaneous, so the sums of their occurrences for thanatin (36.9 + 51.2, Table 5) and for thany2 (45.4 + 41.6, Table 5) result in a total of 88.1% and 87.0%, respectively. These values are very close. The case of the HB between Thr15 and Asn12 is different since the reduction of its occurrence occurs because the Asn12 residue forms an HB with the Lys17 residue in thany2, which does not take place in the thanatin peptide. The turn connecting the two strands of the β-sheet is composed by six residues (Asn12, Arg13, Arg14, Thr15, Gly16, and Lys17). Mandard et al. [13] reported that this six-membered hairpin conformation is stabilized by an HB between the residues Asn12-Lys17 and by two distorted C = O(i) – NH(i + 3) HBs between the Asn12- Thr15 and the Arg13-Gly16 residues. In the present MD simulations, the same HBs were found, except the one between the residues Arg13-Gly16. However, this HB was found in the simulation of thany2. Therefore, the lack of the disulfide bridge in thany2 was compensated by the increase in the occurrence of the pre-existing HBs and by the new HBs such as the ones between the residues Gly16-Asn13 and Lys17-Asn12. Accordingly, in the present case, the removal of the disulfide bridges and the introduction of tyrosine residues not only essentially preserve almost all the original HBs, but also promote a generalized increase in the HB occurrences, thereby contributing to the stabilization of the β-hairpin structure as a compensation for the lack of the disulfide bridges.
3.3. The Ramachandran Angles
The Ramachandran angles for the three peptides and their mutants were also calculated. Tables 6, 7, and 8 show the maxima in the Ramachandran angle histograms for the pair androctonin/andry4, polyphemusin-I/poly4, and thanatin/thany2, respectively. Experimental data for gomesin [27] assert that both dihedral angles Φ and Ψ for Lys8 are positive, while both Φ and Ψ of the neighbor Gln9 are negative. The same motif was encountered in our previous simulations of gomesin [25], (Φ, Ψ) Lys8 = (+50˚, +75˚), and protegrin-1, (Φ, Ψ) Arg10 = (+55˚, +75˚) peptides. These values correspond to the maxima in the histograms of the Ramachandran angles and not to their averages. Interestingly, both residues occupy the position where the chain starts to fold to form the typical hairpin β-strand. Moreover, in these peptides, some Φ and Ψ angles present two preferential values [25]. These remarks support the premise that such peptides exhibit more than one meta-stable conformation in aqueous solution, which may be correlated with the broad-spectrum of their antibiotic activeties. Actually, the alternative conformations must reflect some flexibility of the structure, which may be responseble for increasing the interaction efficiency between the peptides and different kinds of cellular membrane [25]. The same motif was also observed in the simulations of androctonin, polyphemusin-I, and thanatin. However, the Φ and Ψ angles of the androctonin and thanatin peptides are both negative, while in polyphemusin-I they are both positive. Some residues present a multimodal distribution of the Ramachandran angles, displaying two or more peaks (see Figure 7 for instance), suggesting that, despite the presence of the disulfide bridges, these pep

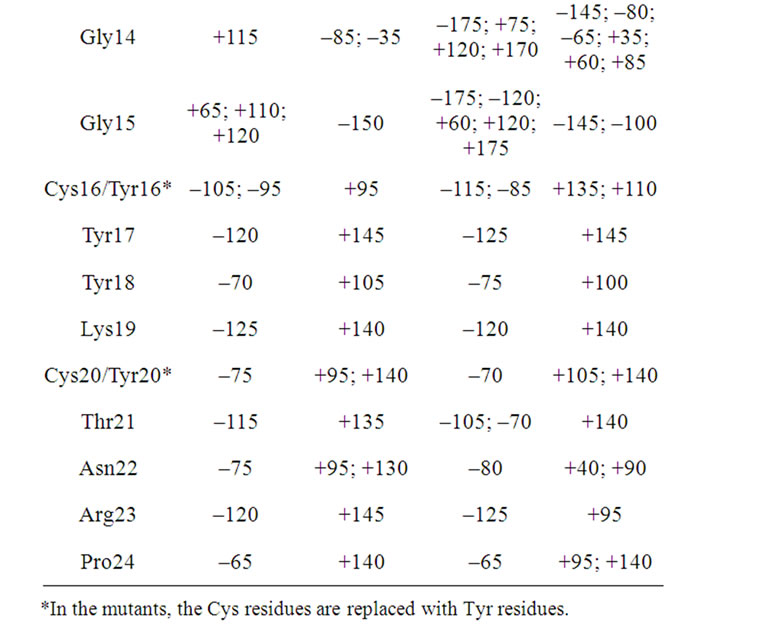
Table 6. The maxima of the Ramachandran angles distributions for the androctonin and andry4 peptides.
tides possess some flexibility, which may be correlated with their broad spectra of action. The maxima of the histograms of the Ramachandran angles for the androctonin Arg12 residue are (Φ, Ψ) Arg12 = (–60˚, –40˚), while in andry4 this motif is observed in the Ser2 residue at (Φ, Ψ) Ser2 = (–115˚, –40˚), Table 6. The polyphemusin-I Arg10 residue presents (Φ, Ψ) Arg10 = (+55˚, +80˚), Table 7. The thanatin Arg13, Arg14, and Thr 15 residues present (Φ, Ψ) Arg13 = (–60˚, –35˚), (Φ, Ψ) Arg14 = (–60˚, –40˚), and (Φ,Ψ) Thr15 = (–95˚, –35˚),


Table 7. The maxima of the Ramachandran angles distributions for the polyphemusin-I and poly4 peptides.

Figure 7. Histograms of the Ramachandran Φ angles for the residues Cys10, Arg11, Arg12, and Arg13 in androctonin.
respectively, while thany2 Ser2 residue has (Φ, Ψ) Ser2 of (–65˚/–120˚, –40˚), Table 8. As reported for the simulations of the gomesin and protegrin-1 peptides [25], the residues number 12 occupy the position where the chain starts to fold. In the case of thanatin, besides the Arg13 residue, where the chain starts to fold, the Φ and Ψ angles of the residues Arg14 and Thr15 are also negative.


Table 8. The maxima of the Ramachandran angles distributions for the thanatin and thany2 peptides.
*In the mutants, the Cys residues are replaced with Tyr residues.
3.4. Interatomic Distances
The distances between the Cα atoms of the cysteine or tyrosine residues were measured for the three peptides and their respective mutants. The average distances between the Cα atoms of the cysteine residues are due to the covalent disulfide bridges linking antiparallel β-strands [28]. The mean distances between the Cα atoms of the cysteine residues in the simulations of the wild peptides are in very good agreement with the experimental ones (mean value in the PDB files), as seen in Table 9. The relatively larger value of the Cα(Cys4)- (Cys20) mean distance in the androctonin peptide, as reported by Mandard et al. [11], allows the formation of

Table 9. Mean distance between the Cα of the Cys residues in the wild peptides and between the Cα of the Tyr residues in the mutants.
the Arg5(CO)-Thr21(NH) HB across the S-S bridge. The Cα(Tyr4)-Cα(Tyr20) average distance (0.59 nm) in andry4 is very similar to the experimental data (0.58 nm) for androctonin. However, the magnitude of the standard deviation (0.06 nm) indicates a considerable fluctuation around the mean value as a result of some flexibility of this region. The Cα(Tyr10)-Cα(Tyr16) average distance (0.44 nm) in andry4 is larger than the corresponding Cα(Cys10)-Cα(Cys16) average distance in androctonin (0.39 nm in experimental data and 0.37 nm in simulation data), showing that this part of the molecule is more separated in andry4 than in androctonin. The distances between Cα found in the simulation of polyphemusin-I are also in good agreement with the experimental ones, considering the magnitude of the standard deviations. The Cα(Tyr4)-Cα(Tyr17) mean distance in poly4 is exactly the same as the experimental value for the Cα(Cys4)-Cα(Cys17) mean distance in the polyphemusin-I peptide, but the standard deviation (0.03 nm) is smaller than the experimental one (0.06 nm), indicating that polyphemusin-I has more flexibility in this region compared with poly4. The Cα(Tyr8)-Cα(Tyr13) mean distance is slightly larger than the corresponding Cα(Cys8)-Cα(Cys13) mean distance in polyphemusin-I, showing that the two sheets in poly4 are more separated than those in the same region in polyphemusin-I. The thanatin Cα(Cys11)-Cα(Cys18) mean distance (simulation data) is very close to the experimental data. The withdrawal of the disulfide bridge in the thany2 peptide causes a small increase in this distance (0.43 nm). All these results suggest that the mutants present good structural similarities with the respective wild peptides. However, the differences in the Cα distances suggest the wild peptides and the mutants do not have exactly the same conformations.
In order to obtain more information about the differences in the secondary structures of the mutants and of the respective wild peptide, some Cα distances between neighbor residues were measured. Large values could mean a loss of the original β-hairpin structure. Figure 8 shows the distributions of the Cα distances for neighboring residues in the androctonin and andry4 peptides (plot A and plot B, respectively). Two kinds of profiles can be observed: one kind with a set of curves between 0.30 and 0.40 nm and a second group with a set of curves appearing between 0.50 and 0.55 nm (Figure 8(a)). This pattern of two kinds of distributions results from the androctonin β-hairpin structure, which is anti-parallel. The same pattern can be observed for andry4 in Figure 8(b), indicating that the mutant does not lose the anti-parallel structure. However, it can be seen that the extremities of the molecule are more separated in andry4 compared with androctonin, as can be seen in Figure 8(b) by the shift to larger values of the distribution curves of the Cα(Ty4)-Cα(Asn22) and Cα(Arg5)-Cα(Thr21) distances (black and red lines, respectively). The distance between the Cα(Tyr10)-Cα(Tyr16) residues in andry4 is larger than that between the corresponding Cα(Cys10)- Cα(Cys16) residues in androctonin (violet line in Figure 8), due to the replacement of the Cys residues with Tyr residues. Once these residues in andry4 do not form an HB, the removal of the disulfide bridge allowed for a slightly increase in the Cα distance. The Cα(Ile7)- Cα(Lys19) distance in andry4 (Figure 8(b) yellow line) is slightly smaller than in androctonin because of the deletion of the disulfide bridges in andry4, which promotes an increase in the HB occurrences shown in Table 3 (82.3% and 85.8% for androctonin, and 87.3% and 90.0% for andry4), thereby resulting in the approximation of these residues. Figure 9 presents the distributions of the Cα distances of neighbor residues in the polyphemusin-I and poly4 peptides (plots A and B, respecttively). Here we also observe two kinds of profiles where one set of curves is found between 0.30 and 0.40 nm and a second set of curves appears between 0.50 and 0.60 nm (Figure 9(a)). The distances between Cα(Tyr4)- Cα(Tyr17) and Cα(Tyr8)-Cα(Tyr13) in poly4 (Figure 9(b), black line, and cyan line respectively) are a little bit larger compared with the corresponding distances Cα(Cys4)-Cα(Cys17) and Cα(Cys8)-(Cys13) in the polyphemusin-I peptide (Figure 8(a), black line and cyan line respectively). The removal of the disulfide bridges and the absence of HB between these residues in poly4 allow these residues to be a little more distant from each other, resulting in small conformational changes. The distance between Cα(Tyr9)-Cα(Phe12) decreases. In this

Figure 8. The distributions of the Cα distances of neighbor residues: (a) androctonin. r = residue; r4 = Cys4; r5 = Arg5; r6 = Gln6; r7 = Ile7; r8 = Lys8; r9 = I le9; r10 = Cys10; r11 = Arg11; r15 = Gly15; r16 = Cys16; r17 = Tyr17; r18 = Tyr18; r19 = Lys19; r20 = Cys20; r21 = Thr21; r22 = Asn22; (b) andry4. r = residue; r4 = Tyr4; r5 = Arg5; r6 = Gln6; r7 = Ile7; r8 = Lys8; r9 = Ile9; r10 = Tyr10; r11 = Arg11; r15 = Gly15; r16 = Tyr16; r17 = Tyr17; r18 = Tyr18; r19 = Lys19; r20 = Tyr20; r21 = Thr21; r22 = Asn22.

Figure 9. The distributions of the Cα distances of neighbor residues in: (a) polyphemusin-I. r = residue; r4 = Cys4; r5 = Phe5; r6 = Arg6; r7 = Val7; r8 = Cys8; r9 = Tyr9; r12 = Phe12; r13 = Cys13; r14 = Tyr14; r15 = Arg15; r16 = Lys16; r17 = Cys17; (b) poly4. r = residue; r4 = Tyr4; r5 = Phe5; r6 = Arg6; r7 = Val7; r8 = Tyr8; r9 = Tyr9; r12 = Phe12; r13 = Tyr13; r14 = Tyr14; r15 = Arg15; r16 = Lys16; r17 = Tyr17.
case, the absence of the disulfide bridges promoted an increase in the HB occurrence (see Table 4), from 76.5% in polyphemusin-I to 93.3 in andry4, allowing them to get closer. Besides, residues Tyr9 and Phe12 in poly4 form two hydrogen bonds, between Tyr9(NH)- Phe12(CO) and Phe12(NH)-Tyr9(CO). Nevertheless, poly4 continues exhibiting the two sets of curves, characterizing that the mutant does not lose its anti-parallel structure.
Figure 10 shows the distributions of some Cα distances