Congenital Microvillous Atrophy, Report of Two Consecutive Siblings with Complete Histologic, Immunohistochemical and Detailed Electron Microscopic Studies, First Report from Iran ()
1. Introduction
Microvillous inclusion disease (MVD) or microvillous atrophy disorder is a congenital disorder of the small intestinal epithelial cells that presents with persistent and severe diarrhea and it is characterized by enterocyte abnormalities [1] . The diarrhea starts in the first 72 hours of life (early onset form) or in 6 to 8 weeks after birth (late onset) [2] . This inheritance of MVD appears to be autosomal recessive, based on cases occurring in siblings and high incidence of consanguinity [3] . Molecular studies demonstrate mutation of the MYO5B that encoded for myosin 5b has a role in pathogenesis of the MVD [4] . Diagnosis is often delayed because of difficulties in taking a small bowl biopsy specimen in the neonatal period. Light microscopy shows enteropathy, and severe atrophy of the enterocytes brush borders, with instead of, accumulation of Periodic Acid Schiff (PAS) and CD10 positive granules at the apical pole of immature enterocytes [1] . Transmission electron microscopy establishes the diagnosis by identifying abnormal microvillous structures at the luminal border of the enterocytes and apical intra cytoplasmic inclusions lined by microvilli in the same cells [5] . For these children, prognosis is generally poor due to metabolic acidosis with poor compensation. Severe repeated dehydration, infectious and liver disease are caused by parental nutrition [1] . The diagnosis and management of congenital microvillous atrophy is difficult. Early small bowel transplantation gives us hope for treatment of this disease. To our experiment, this disease is very rare in Iran and it is yet unreported, so we decided to report two consecutive siblings with the same disease from Iran.
2. Report of the Cases
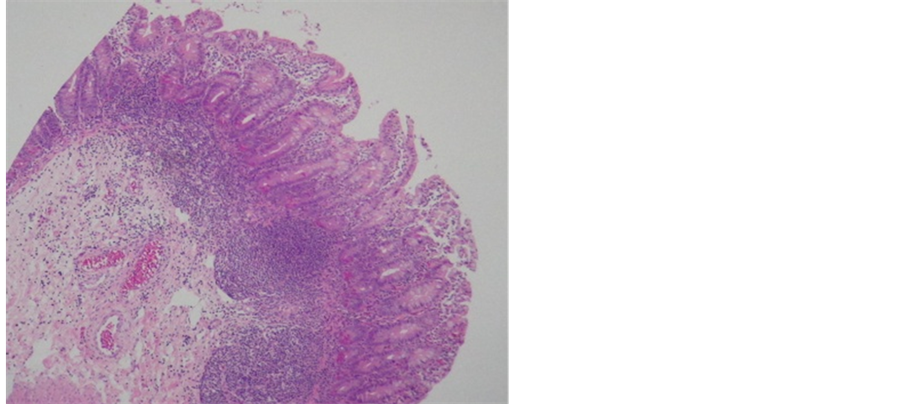
Two siblings (both male) were born to healthy parents who were cousins. The mother had no complications during her pregnancy. Family history is negative for such diseases. The boys were born by cesarean section at the age of 38 and 34 respectively (second boy came after one and half a year). The first child who died at two months of age, had respiratory distress shortly after birth, as a result, surfactant therapy was instituted for him. Both siblings were hospitalized due to severe diarrhea starting shortly after breast feeding. The frequency of diarrhea in both cases was 10 to 17 times per day and their stools were loose and green. The first child was under antibiotic therapy since early in life and received Scholl solution for one day with diagnosis of renal tubular acidosis which was discontinued when diarrhea got worse due to this treatment. There were no other abnormalities in physical examinations. Laboratory data revealed an osmotic diarrhea; no blood or leukocytes were seen in his stool. The first child’s endoscopy was performed 50 days after his birth. Flat villi were seen in duodenum by endoscopy and several biopsies were taken. The second child underwent endoscopy at 10 days of age but biopsy resulted in intestinal perforation, so laparotomy was performed and another full thickness biopsy of small intestine was taken. Histological studies of both siblings revealed duodenal mucosa with complete flattening of villi (total villous atrophy). Superficial lining cells showed atrophy. Crypts showed no hyperplasia, however distortion and difference in size of crypts was apparent (Figure 1). By PAS and CD10 staining, a poorly developed brush border and typical inclusions were seen in apical boarder of enterocytes (Figure 2). Electron microscopy was performed for the second case and showed microvillous involution and inclusions in the apical part of the epithelial cells (Figure 3). Also a normal intestine was studied by electron microscopy as control (Figure 4).
3. Discussion
Persistent and severe diarrhea of infants has a variety of etiologies such as infectious, enzymatic defect, inflammatory conditions, hormonal or metabolic disease and anatomical abnormalities. Despite of extensive investiga-
 (a)
(a)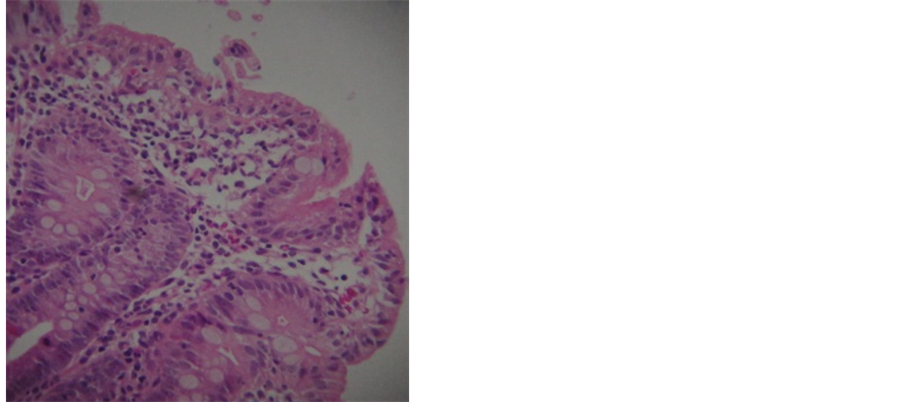 (b)
(b)
Figure 1. (a) Small intestinal mucosa with rather complete flattening of superficial lining but no crypt hyperplasia. Hematoxylin and Eosin stain (×100); (b) There is also mild inflammatory process which is secondary in this case and is not seen in uncomplicated cases of congenital microvillus atrophy. Hematoxylin and Eosin stain (×400).
 (a)
(a) (b)
(b) (c)
(c)
Figure 4. (a)-(c): Electron microscopic appearance of control specimen. Low power and high power, showing well preserved brush border microvilli.
tion, the causes of diarrhea remain unknown in certain cases. Congenital microvillus atrophy, first described in 1978 by Davidson and colleagues, is one of the etiologies of intractable diarrhea in infants. Diagnosis and treatments are difficult. Microvillous congenital atrophy is a rare congenital disorder and it has been estimated that there were no more than a few hundred children with microvillus inclusion disease in Europe. Also some cases with CMA were reported in India, Dutch but based on our information and review of databases, the disease is very rare in Iran and it has not been reported as yet [6] [7] . CMA is a disease with unknown etiology and has no specific treatment except bowel transplantation. Although in some cases partial response to therapy with exogenous epidermal growth factor and the long-acting somatostatin analog octerotide is reported. Due to rareness of CMA, it is crucial to distinguish it from other diseases with persistent and severe diarrhea as soon as possible. Especially due to discovery of new therapies such as small intestinal transplantation alone or in combination with the liver that improves prognosis of these patients and gives us hope for treatment of this disease [8] .
NOTES
*Corresponding author.