Metallated Schiff-Base Macromolecules as Alternative Metalloprotein Electron Transfer Intermediates ()
1. Introduction
Horse heart cytochrome-C presents a very simplistic structural design consisting of 104 amino acids and is often utilized as a prototype for metalloproteinase [1] [2]. Cytochrome-C serves as an electron shuttle, by forming reversible complexes with its redox counterparts during electron transfer [3].
The active site in cytochrome-C is the iron heme that is covalently linked to the enzyme at cys 14 and cys 17 by thioether bonds. During the electron transfer process the redox state of the iron changes between Fe2+/Fe3+.
The iron heme lies in the plain of a porphyrin ring [4], of which the 5th and 6th sites are occupied by the nitrogen of the imidazole ring of histidine. Bonanni et al., (2003) investigated the adsorption of cytochrome-C molecules from yeast on bare gold electrodes [5]. Methodologies for fixing these proteins to an electrode surface without affecting the integrity of the protein made up a major component of the research. The results indicated that yeast cytochrome-C, directly self-chemisorbed onto the gold electrode, retained its morphology and redox properties, with coverage of up to 84% of the electrode substrate. Electron transfer limitations for immobilized cytochrome-C could also be overcome by the application of a monolayer surface modifier [5]. This method promotes electron transfer by orientating the molecules to a fixed and electronically favorable orientation [5].
The electron charge transfer of cytochrome-C is difficult to detect on bare electrodes. Bond et al., (1992) investigated the mass transport mechanisms associated with redox processes of cytochrome-C at gold electrodes modified with 4, 4’-bipyridyl disulphide (SS-bpy). They concluded that modifier layers provide electro active sites under applied potential. They ascribed the improvements to the modifiers unblocking parts of the electrode surface by displacing adsorbed cytochrome-C providing sites for electron transfer [6]. Heat treatment induces functional changes to the structure of the heme of cytochrome-C, His-Fe-Met octahedral coordination and a thioether bond with two cysteine residues of the peptide chain. The peptide chain of hemoglobin or myoglobin bound only the histidine residue as a fifth ligand resulting in unfolding of the peptide chain, to expose the Fe heme as the methionine-sulphur coordination disappeared. Consequently the heme becomes exposed to water, resulting in the heme’s newly acquired autoxidation ability.
Eguílaz et al., (2010) constructed a cytochrome-C based biosensor by drop-coating cytochrome-C on a hybrid glassy carbon electrode modified with poly-(3-methylthiophene) and multiwall carbon nanotubes [7]. The hybrid GCE was coated with a self-assembled monolayer of l-cysteine onto which cytochrome-C was drop coated. The system was evaluated against H2O2, as cytochrome-C type biosensors have been used for the determination of H2O2 and other analytes related to H2O2 quantification [8] [9]. The sensor displayed good performance in the amperometric determination of H2O2. When used in the biocatalytic determination of nitrite the system displayed superior sensitivity.
Electrostatic or chemisorptive end-capping of proteins with gold nanoparticles has been reported to improve distant donor-acceptor electronic coupling [10] [11] [12] [13]. In a study by Caban, Offenhäusser and Mayer (2009) bovine heart cytochrome-C was electrostatically coupled to an atomically flat Au (111) surface through a monolayer of 11-mercaptoundecanoic acid. Binding of the gold nanoparticles to cytochrome-C was identified with AFM to proceed via two mechanisms [14], based on changes in magnitude of the redox response. It was postulated that the attachment of the nanoparticles might have caused substantial deformation of the protein’s ternary structure resulting in increased heme-electrode distance. The redox response of the system was related to immobilization time of the particles and their size. Binding of the Au nanoparticles were found to favorably alter the spatial geometry of cytochrome-C by accelerating the long range electron transfer rate of the protein [14]. Tanimura et al., (2002) reported on modifying the electrode surface with a self-assembled monolayer of alkanethiols. The positively charged lysine residues subsequently aligned with the negatively charged carboxylate head group, enhancing the electron transfer capabilities [15].
In this work, we endeavor to reproduce the redox chemistry of the iron heme of denatured cytochrome-C whilst circumventing the complexities associated with functionalizing cytochrome-C for sensor application. The heme structure was emulated by synthesizing a Schiff-base ligand and complexing it with iron. The electron transfer properties of the macrocycle were evaluated by means of voltammetry and compared to that of commercial denatured cytochrome-C modified Schiff-base.
2. Experimental Section
Starting materials, 2, 3-diaminonaphthalene and pyrrole-2-carboxaldehyde, were purchased from Sigma Aldrich and used without further purification. Propanol (Sigma) was dried over molecular sieves at room temperature. Dimethylformamide and 37% HCl was purchased from KIMIX chemicals. 0.1 M HCl and 0.1 M NaBF4 supporting electrolyte solutions were prepared with Millipore deionized water from a Millipore Synergy water system with a resistivity of 18 MΩ.
The Schiff-base was synthesized through the condensation reaction between 2, 3-diaminonaphthalene and pyrrole-2-carboxaldehyde under anhydrous conditions in propanol. The reaction was initiated by dissolving the amine, 2, 3-diaminonaphthalene, in dried propanol. In a separate vessel pyrrole-2-carboxaldehyde was dissolved in dried propanol and heated prior to addition to ensure dissolution. The dissolved aldehyde was added drop-wise to the amine. The reaction was performed under reflux at 60˚C. The reaction progress was monitored with thin layer chromatography on silica plates in 90:10 ratio hexane ethanol mobile phase. At hour 72 the reaction reached completion [16].
Schiff-base reactions are nucleophilic substitution reactions which are generally either acid, base or heat catalyzed. The acidic nature of the aldehyde pyrrole-2-carboxaldehyde negated the need for additional acid catalysis. Acidic catalysis activate the carbonyl carbon for nucleophilic attack. The 2, 3-diaminonaphthalene acts as a weak nucleophile due to steric hindrance. The carbinolamine produced is a tetrahedral intermediate and unstable, which undergoes acid catalyzed dehydration [17]. Condensation reactions are trademarked by the production of water, which in turn impacts on reversibility of the reaction. Therefore it was imperative to keep reaction conditions free of water.
H1NMR spectra were recorded on a Bruker BioSpin GmbH 300 in CDCl3. FTIR analysis was performed on PerkinElmer spectrophotometer over the range of 400 to 4000 cm−1. 4 mg sample aliquots were mixed with dried KBr and compressed into pellets with a PerkinElmer KBr pellet quick press. Starting materials and ligand product were prepared as 100 µg/L samples for UV/Vis absorbance, measured on a Nicolet Evolution 100 UV/Vis spectrophotometer, over the 200 to 900 nm spectral range with 10 mm pathlength Hellma absorption cuvettes in ethanol.
Electron Microscopy, for EELS analysis, required FeCl2∙4H2O and Fe-Schiff-base complex to be coated directly onto Cu stubs and sputter coated with activated carbon. Analysis was performed on a new Field Emission Tecnai F20. High resolution SEM coupled to EDS analysis was performed on a Zeiss Auriga EMU.
Electrochemical experiments were performed on the PalmSens potentionstat using DropSens screen-printed carbon electrodes. The ligand and the Fe-Schiff-base complexes were electrodeposited onto the screen printed carbon electrodes with cyclic voltammetry in the range −0.6 V to 0.7 V (vs. Ag/AgCl) for five cycles. The stability of the Fe-Schiff-base complex was evaluated by cycling in phosphate buffer electrolyte solution pH 7, from −1 to 1 V at scan rates 10, 50 and 100 mVs−1 (n = 3). Spectrometric analysis was conducted on all three electrolyte solutions to identify leaching of iron into the solution
3. Results and Discussion
Schiff-base synthesis. The Schiff-base N, N-bis (1H-pyrrole-2yl) methylene naphthalene-2, 3-diamine was synthesized by the condensation reaction of 2, 3-diaminonaphthalene and the acidic aldehyde pyrrole-2-carboxaldehyde (see Scheme 1).
The H NMR spectrum showed an absence of the characteristic amine and carbonyl chemical shift values expected for 2, 3 diaminonaphthalene and pyrrole-2-carboxaldehyde respectively. The spectrum of the Schiff-base produced showed two symmetrical peaks i.e. at 8.1 and 7.7 ppm which was evidence of
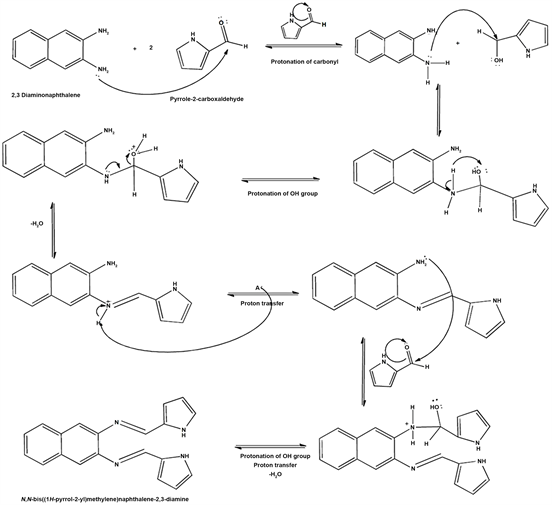
Scheme 1. Proposed mechanism for the preparation of Schiff-base ligand N, N-bis (1H-pyrrole-2yl) methylene naphthalene-2, 3-diamine.
imine formation (-N=CH-). These chemical shift values for the imine coincided with reported data by Simionescu et al., (1998) who synthesized and polymerized pyrrolylidene naphthylamine Schiff-base compounds.
Infrared analysis was performed on the starting materials as well as Schiff-base product (see Table 1). The spectra identified characteristic absorption bands for each compound. The FTIR spectrum of Schiff-base produced molecular vibrations and rotations that were a combination of 2, 3-diaminonaphthalene and pyrrole-2-carboxaldehyde. 2,3-Diaminonaphthalene identified an absorption band at 3380 cm−1 which coincided with reports in literature for primary amines [18]. The absorption band at 1595 cm−1 coincided with literature reports and confirmed the formation of the Schiff-base through the formation of the imine bond [19] [20]. The shift in the wavelength coincided with findings in literature. The spectra of the ligand also indicated the absence of the C=O shift at 1633 cm−1 [16]. Extending pi conjugation has been reported by Wei et al., (2002) to result in a red shift. The study investigated the effect of pi conjugation on the properties of boron difluoride [21]. A theoretical study by Bonanni et al., (2003) on the effects of substituents, such as cyano and carbonyl groups on pyrrole, found these groups to have an effect on the HOMO and LUMO levels [22]. The groups were found to decrease the band gap and increase the conjugation length of the polymers which impacts absorbance. In the spectra of the Schiff-base similar shifts in stretching vibrations were evident for the pyrrole ring.
![]()
Table 1. FTIR bands relevant to N, N-bis (1H-pyrrole-2yl) methylene naphthalene-2, 3-diamine characteristics.
Distinctly different UV spectra were obtained for pyrrole-2-carboxaldehyde, 2, 3-diaminonaphthalene and Schiff-base product. For the product three bands were detected at 230, 288 and 349 nm. The bands at 230 nm and 288 nm were attributed to the π-π* transitions of the aromatic rings of pyrrole and naphthalene. The absorption band at 349 nm was attributed to the π-π* of the imine [23] (Figure 1).
When the spectrum of the product was compared to the starting materials changes in the peak height, shape and size was evident. Peak broadening was observed for the Schiff-base that was evidence of extended conjugation. The absorption band in the region of 275 to 300 nm for the Schiff-base was assigned to n-π transition in the aromatic rings. The absorption band at 350 nm for the Schiff-base was assigned to π-π* in the aromatic ring and C=N [20] [24]. The diaminonaphthalene starting material peak recorded at 250 nm was absent in the Schiff-base product.
Infrared analysis was performed on the starting materials and the monomer, and compared as per Figure 2. The results identified a spectra with characteristic absorption bands for each compound. N, N-bis(1H-pyrrole-2yl)methylene naphthalene-2,3-diamine identified molecular vibrations and rotations that were a combination of 2,3-diaminonaphthalene and pyrrole-2-carboxaldehyde. 2,3-Diaminonaphthalene identified an absorption band at 3380 cm−1 which coincided with reports in literature for primary amines [18]. The absorption band at 1595 cm−1 coincided with literature reports, and confirmed the formation of the Schiff-base which was characterized by formation of an imine bond [18] [19] [20]. The shift in the wavelength coincided with findings in literature. Extending pi conjugation has been reported by Barbon, Staroverov and Gilroy (2015) resulted in red shifts [21]. The study investigated the effect of pi conjugation on the properties of boron difluoride. The study found pi conjugation to have a direct impact on wavelength absorption which resulted in a red shift. Isaiah, Adejoro and Collins (2013) found substituents, such as cyano and carbonyl groups on pyrrole, to have an effect on the HOMO and LUMO levels [22]. Subsequently, the Schiff-base product was used as the ligand for complexation with Fe under reflux.
Fe complex synthesis: The macromolecule was synthesized by refluxing a 5 mM methanolic solution of the metal precursor [FeCl2∙4H2O] in a 10 mM methanolic solution of the monomer. The solution was heated to 60˚C and
![]()
Figure 1. UV/Vis absorbance of starting materials 2, 3-diaminonaphthalene, pyrrole-2-carboxaldehyde and synthesized product N, N-bis (1H-pyrrole-2yl) methylene naphthalene-2, 3-diamine (Schiff-base) in ethanol.
![]()
Figure 2. FTIR spectra of starting materials 2,3-diaminonaphthalene, pyrrole-2-carboxaldehyde and monomer N, N-bis(1H-pyrrole-2yl)methylene naphthalene-2,3-diamine in dried KBr.
refluxed for three hours after which a dark coloured precipitate was obtained. At the 30 minute mark there was a colour change of the solution from red brown to black. Scheme 2 predicts a possible mechanism and structure for the macromolecule based on the iron chelating and ligand binding effect of N,N-bis(1H-pyrrole-2yl)methylene naphthalene-2,3-diamine. Pyrrole in a study by Doddi et al., (1976), who investigated the reactivity of pyrrole in nucleophilic
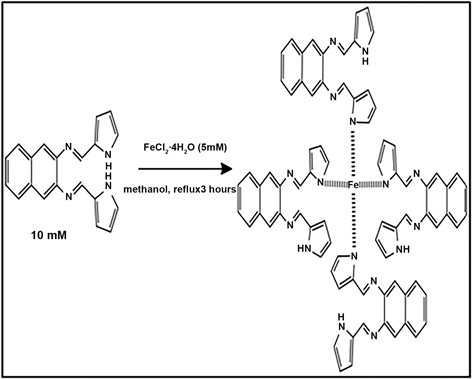
Scheme 2. Proposed mechanism for the preparation of the macromolecule.
substitution in comparison to furan and thiophene derivatives, found pyrrole to be not as reactive relative to the other heteroatoms [25]. Ligands with soft-donor atoms prefer iron (II), such as nitrogen containing ligands [26]. This predicate suggested the metal to ligand binding between the pyrrole lone pair and iron. The higher the number of chelating rings attached to the chelator the higher the stability [26]. A square planar coordination with four ligands chelated to Fe was proposed. It offered substantial electronic stabilization. Strong field ligands have been reported to adopt square planar geometry in Ni complexes. Square planar coordination for iron (II) compounds have been reported to be limited to complexes containing porphyrin, salicylaldimato, phthalocyanine ligands [27]. Based on the premise of N, N-bis (1H-pyrrole-2yl) methylene naphthalene-2, 3-diamine being a heterocyclic macrocycle organic compound it was deduced that the macromolecule would adopt square planar geometry.
FTIR analysis of the Fe-Schiff-base ligand complex is presented in Figure 3. The spectra showed a shift in frequency for some ligand bands and also identified a new band 473 cm−1 which was attributed to metal ligand complexation [28] [29] [30]. This band was absent in the unbound ligand. The azomethine (imine bond) vibrational stretch was recorded at 1595 cm−1, for the ligand. According to literature a shift toward lower frequency would have been recorded if the iron complexed through the azomethine nitrogen, and a shift to higher frequency would indicate shortening bond length [31] [32] [33]. Evidence suggests that the metal ligand coordination did not proceed via the imine linkage, since a shift to higher frequency was recorded for the azomethine group at 1630 cm−1. This shift confirmed that the iron did not complex with the azomethine group and complexation occurred at the pyrrole nitrogen. The broad peak at 3200 cm−1 was evidence that one of the 2 pyrrole units retained an uncomplexed nitrogen, for each ligand binding to the Fe centre [32].
![]()
Figure 3. FTIR spectra of the Schiff-base ligand and Fe-Schiff-base ligand complex prepared as dried KBr pellets.
The UV/Vis spectra of the Fe-Schiff-base ligand complex was measured and compared with that of the ligand N, N-bis (1H-pyrrole-2yl) methylene naphthalene-2, 3-diamine as presented in Figure 4. The Fe-Schiff-base ligand complex produced three absorption peaks at 225, 287 and 350 nm, respectively. The peaks at 225 nm and 287 nm were attributed to the π-π* transitions in the aromatic rings of pyrrole and naphthalene. The absorption band at 350 nm can be attributed to the π-π* of the imine. When the spectra of the Fe-Schiff-base ligand complex was compared to the ligand a narrowing of the bands were evident. These changes were attributed to the coordination with iron. The spectrum identified weak absorbance above 400 nm which could be the d-d transitions of the metal but the intensity of the transitions were too weak to be conclusive on its own [34].
Transition electron microscopy (TEM) was performed on the ligand and the Fe-Schiff-base ligand complex. The samples were sputter coated onto copper grids for analysis.
In Figure 5 and Figure 6 the high resolution TEM micrographs shows the structural morphology of the ligand (Figure 5) and Fe-Schiff-base ligand complex (Figure 6). The morphology of the Fe-Schiff-base ligand complex (Figure 6) appears more structured following chelation with iron. In order to confirm successful coordination of iron to the monomer (N, N-bis (1H-pyrrole-2yl) methylene naphthalene-2, 3-diamine) electron energy loss spectroscopy (EELS) was performed on the Fe-Schiff-base ligand complex as well as the starting material FeCl2∙4H2O. The EELS spectrum in Figure 7 of the Fe-Schiff-base ligand complex provided an absolute measurement of iron in the complex. Transition metals exhibit L3 and L2 edges which is an expression of the excitations of electrons from 2p to unfilled 3d orbitals. The orbital transitions are represented by ratios.
![]()
Figure 4. UV/Vis spectroscopy of the N, N-bis (1H-pyrrole-2yl) methylene naphthalene-2, 3-diamine and Fe-Schiff-base ligand complex in ethanol 1 mg/ml 100× dilution.
![]()
Figure 5. TEM micrographs of the ligand.
![]()
Figure 6. TEM micrographs of Fe-Schiff-base ligand complex.
![]()
Figure 7. EELS analysis of FeCl2∙4H2O (black curve) vs Fe-Schiff-base ligand complex (red curve).
Variations in L3/L2 intensity ratios are related to changes to the electronic structure of the d orbitals. In order to monitor changes in the L3/L2 intensity ratios of the iron EELS was performed on Fe-Schiff-base ligand complex as well as the iron precursor (see Figure 7). Analysis of the results revealed a change in the peak ratios. Changes to the peak ratios would be evidence of a change in electron density surrounding the iron [35] [36]. Based on these findings coordination of iron to the monomer was confirmed. EELS also revealed iron to remain in the Fe2+ oxidation state with almost similar energy-loss spectra to that of FeCl2∙4H2O.
EDS analysis was performed on the Fe-Schiff-base ligand complex to confirm the presence of iron in the complex (see Figure 8 and Figure 9). The results showed no iron was present in the ligand (Figure 8) but present in the Fe-Schiff-base ligand complex (Figure 9).
Electrochemical experiments. Denatured cytochrome-c (1 mg/mL stock solution) was electrodeposited directly onto a bare SPCE by cycling from −0.600 V to 0.700 V at 50 mV/s. The Schiff-base ligand was electrodeposited onto a SPCE in the same way, starting from a 5 mM equivalent of the ligand solution, prepared by dissolving (5 mM) N, N-bis(1H-pyrrole-2yl) methylene naphthalene-2,3-diamine dissolved in dimethylformamide 0.1 M NaBF4 mixed electrolyte system. Cyclic voltammetry was used to deposit the ligand onto the working electrode, by cycling the potential between −600 mV to 700 mV for 5 cycles.
Scan rate dependent studies were done using cycling voltammetry varying the scan rate from 10 mV/s to 500 mV/s in the potential window −1 V to 1 V.
In the forward scan a non-reversible oxidation process was recorded producing a peak at 0.687 V vs Ag/AgCl (Figure 10). The oxidation process at 0.687 V was a one electron transfer process, electrochemical in nature, that produced Ph=[NHC5H7N]+, an unstable intermediate [34] [35] [37]. The anionic radical,
![]()
Figure 8. Energy-Dispersive X-ray Spectroscopic (EDS) analysis of Schiff-base ligand.
![]()
Figure 9. Energy-Dispersive X-ray Spectroscopic (EDS) analysis of Fe-Schiff-base ligand complex.
was reduced on the reverse scan and a strong reduction peak was observed at −0.676 V Vs Ag/AgCl in the cathodic scan [38]. At 10 mV/s the peak to peak
![]()
Figure 10. Cyclic voltammetry of SPCE modified with Schiff-base ligand, in 0.1 M PBS vs Ag/AgCl electrode at scan rates 10 mV/s to 500 mV/s in the potential window −1 to 1 V.
separation was calculated at ∆E = 1.36 V and the electron transfer process was determined to be non-reversible.
At higher scan rates the electron transfer in the system changed from non-reversible to poorly defined quasi-reversible, based on peak separation values. The peak current behavior in response to changes in scan rate indicated adsorption behavior during the oxidation waves and diffusion controlled electron transfer during the reduction waves, recorded [39].
Denatured cytochrome-C was coated onto a bare SPCE (Figure 11(a)) and compared to cytochrome-C coated onto the Schiff-base ligand modified SPCE (Figure 11(b)). An irreversible anodic peak was recorded at 0.420 V for denatured cytochrome-c deposited onto the bare SPCE (Figure 11(a)) which was attributed to Fe oxidation in denatured cytochrome. Lysine has been reported to facilitate redox processes within the enzyme but high temperature denaturation deprotonates lysine. This results in changes in the redox behaviour of the enzyme [40] [41]. For the ligand modified SPCE onto which denatured cyto-chrome-C was drop coated, a quasi-reversible redox couple was recorded at −0.524 V vs Ag/AgCl at 10 mV/s consistent with Fe2+/Fe3+ redox behaviour in macromolecules. These findings coincided with reports in literature. Sagara et al., (1990) found electron transfer reactions to be facilitated by surface modifiers in their investigation of the behaviour of cytochrome-C deposited on gold electrodes modified with 4-pyridyl derivatives [42]. Similar findings were reported by Eddowes and Hill (1979), when investigating the impact of monolayers on the electron transfer behaviour of cytochrome-C. The ligand modified SPCE drop-coated with denatured cytochrome-C (Figure 11(b)) displayed oxidizing potentials more negative than denatured cytochrome-c on bare SPCE with reversible electron transfer behavior (Figure 11(a)).
Diffusion controlled electrochemical processes, typically follow Randle-Sevҫik
![]() (a)
(a)![]() (b)
(b)
Figure 11. (a) Cyclic voltammetry of SPCE modified with denatured cytochrome-C (b) Ligand modified SPCE onto which denatured cytochrome-C was drop-coated in 0.1 M PBS vs Ag/AgCl electrode at scan rates 10 mV/s to 500 mV/sin the potential window −1 to 1 V.
kinetics according to equation,
, where (A) is
the surface area, (De) diffusion coefficient and (C) analyte concentration. The systems displayed diffusion controlled kinetics as evidenced by the linear regression lines for scan rates 10 - 500 mV/s. The diffusion coefficient (De) was calculated to be 1.07 × 10−6 cm2/s for the anodic process and 1.31 × 10−6 cm2/s for the cathodic process. These values were in agreement with a report by Eddowes and Hill (1979). De values of 9.4 × 10−7 cm2/s were reported for 4,4'-bipyridal cytochrome-c systems [43]. Abass and Hart (2001) reported De values of 8.1 × 10−7 cm2/s for the anodic peak currents and 8.03 × 10−7 cm2/s for the cathodic peak currents in their investigations of cytochrome-c deposited on polycarbonate modified SPCE [44].
Cyclic voltammetry was performed on the SPCE modified with the Fe-Schiff-base ligand complex. Cyclic voltammetry showed the system to be electroactive (Figure 12) in the electrochemical window −1 V to 1 V vs Ag/AgCl. A well-defined redox couple was recorded at −0.429 V vs Ag/AgCl. Coordination of Fe to the ligand system appeared to have shifted the potential at which oxidation and reduction occurred. In the forward scan an anodic peak was recorded at −0.414 V and cathodic peak at −0.507 V in the reverse scan.
Reference values for uncomplexed iron, are given as [45].
and
However the values for Fe oxidation and reduction, when iron is complexed within a macromolecule structure, changes. Negative shifts in redox potentials, according to reports, is an indication of increased stability of Fe [46] [47]. This was highlighted for Fe in cytochrome-C in this work, and the redox potentials observed for the Fe-Schiff-base ligand complex was in good agreement with the redox potentials measured for cytochrome-C electrodeposited at the SPCE electrode, in the same way.
Diffusion coefficient values for the Fe-Schiff-base ligand complex deposited onto the SPCE were 8.86 × 10−7 cm2∙s−1 for oxidation currents (Ipa) and 9.38 × 10−7 cm2∙s−1 for reduction currents (Ipc). Compared to phenazine-2, 3-diimino (pyrrole-2-yl) (PPDP), a similar type hinge system, doped with 1, 4-naphtaquinone sulphonic acid (NQSA) and polyvinylsulfonic acid (PVSA) the diffusion rates were competitive. The following diffusion coefficient values for PPDP doped with NQSA were reported; De of 4.62 × 10−7 cm2∙s−1 (Ipa) and 4.39 × 10−7 cm2∙s−1 (Ipc). PDDP doped with PVSA on the other hand reported De values 9.28 × 10−3 cm2∙s−1 (Ipa) and 1.60 × 10−2 cm2∙s−1 (Ipc). Zaheer et al., (2011) investigated the introduction of ferrocene moieties into Schiff bases with De values of 2.66 × 10−5 cm2/s−1 [46].
![]()
Figure 12. Cyclic voltammetry of SPCE modified with Fe-Schiff-base ligand complex in 0.1 M 0.1 PBS vs Ag/AgCl electrode at scan rates 10 to 500 mV over the potential window −1 to 1 V.
Comparing the diffusion coefficient values for the ligand, ligand modified with denatured cytochrome-C and Fe-Schiff-base ligand modified SPCEs, the presence of iron significantly impacted the electron transfer characteristics of the Schiff-base complex.
Stability of Fe-Schiff-base ligand complex modified electrode. The stability of the iron within the Fe-Schiff-base ligand complex modified electrode, under redox conditions, was evaluated. The Fe-Schiff-base ligand complex, deposited onto a SPCE, was connected as the working electrode in a 3 electrode arrangement and subjected to potential cycling in 0.1M PBS. CV analysis was conducted at three different scan rates 10 mV/s, 50 mV/s and 100 mV/s within the potential window −1 V to 1 V. UV/Vis absorbance measurements were subsequently performed on the electrolyte solutions, after each CV scan in order to identify the presence of Fe in the electrolyte solution as evidence of possible leaching of complexed iron during redox cycling. This experiment was performed in triplicate and labelled A, B and C.
Electrochemical analysis showed no changes to the redox chemistry of the Fe-Schiff-base ligand system (see Figures 13-15). The forward scan recorded an anodic peak current at −0.566 V and cathodic peak current at −0.636 V in the reverse scan. The cyclic voltammetric results showed no evidence of iron leaching into the solution.
UV/Vis absorbance confirmed the presence of Fe, only, after the initial scan (see Figure 16). However, in subsequent analysis no evidence of leaching could be observed. Upon continued cycling the subsequent electrolyte solutions were found to be free of Fe. The presence of Fe initially observed in the UV/Vis data was assigned to loosely bound residual material from the electrodeposition process.
![]()
Figure 13. Cyclic voltammograms of the Fe-Schiff-base ligand complex electrodeposited onto a screen print carbon electrode in PBS at; performed at 10 mV/s for scans A, B and C.
![]()
Figure 14. Cyclic voltammograms of the Fe-Schiff-base ligand complex electrodeposited onto a screen print carbon electrode in PBS at; performed at 50 mV/s for scans A, B and C.
![]()
Figure 15. Cyclic voltammograms of the Fe-Schiff-base ligand complex electrodeposited onto a screen print carbon electrode in PBS at; performed at 100 mV/s for scans A, B and C.
![]()
Figure 16. UV/Vis analysis of PB Selectrolyte after potential cycling, for each of the respective scan rate experiments.
4. Conclusion
A novel heterocyclic metal conjugated Schiff-base was synthesized and studied with respect to its structure and electron transfer capabilities. Naphthalene diamine was found to be a suitable nucleophile for the Schiff-base synthesis in spite of its bulky nature. The Schiff-base was further successfully complexed with Fe to produce the Fe-Schiff-base ligand complex. Cyclic voltammetry confirmed that the electron transfer properties of denatured cytochrome-C deposited onto a Schiff-base ligand modified SPCE, were reproduced in the Fe-Schiff-base ligand complex electrodeposited directly onto the SPCE. The Fe-Schiff-base ligand complex displayed electron transfer properties similar to that of the denatured metalloprotein, studied in this work and presented a stable alternative for heme facilitated sensing applications, without the restriction of physiological pH conditions during application of the sensor to a sample solution.
Acknowledgements
The authors wish to acknowledge the National Research Foundation (South Africa) for research funding.