Molecular Modelling of Selected Combustion By-Products from the Thermal Degradation of Croton megalocarpus Biodiesel ()

1. Introduction
Transport fuel emissions from fossil fuels have compromised air quality, affected human health for decades, and caused numerous health miseries [1]. Consequently, biomass fuels have gained enormous attention lately because they are promising renewable energy resources. These fuels have proven to reduce greenhouse gas emissions and other environmental pollutants. From recent studies carried out by [2], it is evident that the ratio of biodiesel to fossil diesel yields better combustion results which decrease the yield of toxic pollutant emissions. Biodiesel emissions have been known to be free from harmful aromatic compounds as opposed to commercial diesel [3]. This is beneficial in the combustion efficiency of biodiesel which has been noted to suffer from low oxidation ability, increased acidity and corrosion problems [4]. Environmentally persistent pollutants are considered stable and spend long lifetimes in the environment. Understanding the reactivity and stability of these pollutants is of the essence to public health practioners and health policy authorities. One of the key factors controlling the physical properties of substances is the band gap energy, which forms the primary focus of this study. Furthermore, bond dissociation energies of these compounds are another determinant in the interpretation of the reactivity and stability of molecular compounds.
The complicated structural composition of biomass materials makes it difficult to link formation of organic pollutants to exact chemical reactions but with the use of model compounds it is possible to understand the structural entity of biomass components of significant importance in the transport industry and environmental health [5]. Hitherto, the world today is witnessing serious concerns regarding the depletion of petroleum reserves and environmental degradations because of hazardous emissions resulting to severe health consequences [6]. Biodiesel has received significant attention as an environmental friendly fuel chiefly composed of fatty acids, methyl esters derived from renewable feedstock, perceived as potential substitutes to petro-diesel for engine operations [7].
The thermal decomposition of fuels leads to the generation of smaller fragments of decomposition of biodiesel blends such as transient radicals and stable species which result in the evolution of CO2 (decarboxylation reaction), olefins, water, aldehydes, ketones, and small alkanes such as methane, ethane and propane [8] [9]. Radicals formed from the thermal degradation events have generally been referred to as environmentally persistent free radicals (EPFRs) and represent a class of reactive species that have prolonged lifetime even under ambient environmental conditions [9]. Radical stabilities and reactivity can be explored by the use of computational techniques as opposed to experimental methods due to their very short half-lives. A research carried out by Kibet and co-workers (2018) has shown that some of the radicals generated by the thermal decomposition of biodiesel blends have long half-life ≈ 431 days. This research output shades more light on the existence of environmentally persistent free radicals (EPRs) in the environment originating from combustion events. This study focuses on examining the physical properties of some selected volatile organic emissions from the co-pyrolysis of biodiesel blends with inclination to electronic properties.
Computational chemistry provides understanding into the molecular properties of a compound that would not otherwise be easy to determine experimentally. Additionally, computational methods are economical in terms of cost and time. This study investigates in detail the electron density maps, molecular orbitals, band gap energies and geometry optimization of selected combustion by-products of Croton megalocarpus bio-oil; 4-(2,4-dimethylcyclohexyl)-2-butanone, 3,4-dimethyl-3-cyclohexene-1-carbaldehyde, 5-(3-phenylpropanoyl)dihydro-2-(3H)-furanoneand isopropenyl-4-methyl-1,2-cyclohexanediol. The electron density contour maps generated was used to examine the nucleophilicity of the selected volatile organic compounds in order to gain more insight on how they interact with biological structures to cause toxicity and cellular impairment in respiratory systems. The optimization process for various pathways for the 5-(3-phenylpropanoyl)dihydro-2(3H)-furanone and intermediate free radicals was probed. Moreover, the thermochemistry and molecular models for the various mechanistic pathways for radical formation and their conversion to free molecules were proposed.
2. Methodology
Density Functional Theory (DFT) Calculations
DFT calculations were performed by use of Gaussian ‘16 computational software [10]. Geometry parameters such as compound energetics and bond energies were calculated at the ground state geometries employing B3LYP level of theory with 6-31G basis sets. To develop molecular orbital energy-level diagrams, electronic density contour maps and the determination of the band gap energy for selected molecules, Gaussian 16 software outputs were interfaced with Chemissian version 4.43 [11]. All computational calculations were conducted at 298.15 K and 1 atmosphere. To compute the energy change for formation of a compound or a free radical from its components, the following thermodynamic equation (Equation (1)) was used [12].
(1)
where,
is change in enthalpy of the reaction,
is correction to the thermal enthalpy and
is the sum of electronic and thermal enthalpies
3. Results and Discussion
3.1. Molecular Geometries of Major Molecular Emissions
Geometry parameters such as bond lengths and bond angles have a significant influence on the strength of the bonds of molecular structures. The structures of selected model components are presented in Scheme 1, vide infra. The
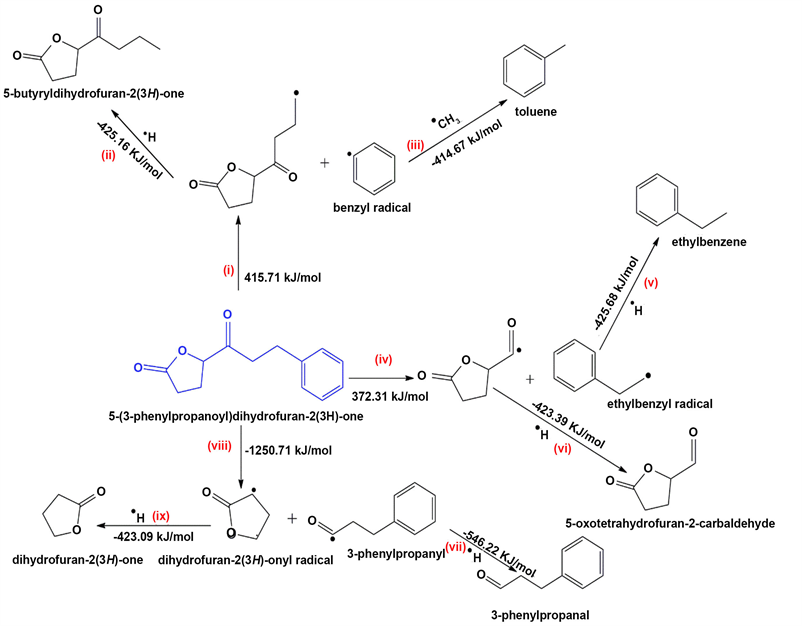
Scheme 1. Proposed mechanistic pathways for the thermal degradation of 5-(3-phenylpropanoyl)dihydro-2-(3H)-furanone.
optimization process presented below in Figures 1-4 show steps towards achieving an optimized Structure for 4-(2,4-Dimethylcyclohexyl)-2-butanone, 3,4-Dimethyl-3-cyclohexene-1-carbaldehyde, 5-(3-phenylpropanoyl)dihydro-2(3H)-furanoneand isopropenyl-4-methyl-1,2-cyclohexanediol. This offers interesting quantum mechanics results that cannot easily be accomplished by experimental methods.
The optimization of 3,4-dimethyl-3-cyclohexene-1-carbaldehyde proceeds 31 steps to attain a structure of zero energy (global minimum structure).
Interestingly, 5-(3-phenylpropanoyl) dihydro-2(3H)-furanone (Figure 2) required 91 steps to attain a global minima structure. This is attributed to its large molecular weight of 218.09 g/mol. The scission of C5-C6 (cf. Figure 3) bond in 5-(3-Phenylpropanoyl) dihydro-2(3H)-furanone proceeds with a bond dissociation energy of 372.31 kJ/mol. This was one of the thermodynamically feasible pathways (Scheme 1, route (i)) becauseit proceeds with lower energy compared to the rest of channels. The scission of C4, C5 and C7, C8 proceed with energy barriers of −1250.75 and 415.71 kJ/mol, respectively.
![]()
Figure 1. Optimized structure of 3,4-dimethyl-3-cyclohexene-1-carbaldehyde and a plot showing its optimization steps (the red circle is the optimization level).
![]()
Figure 2. Optimized structure of 5-(3-phenylpropanoyl) dihydro-2-(3H)-furanone and a plot showing its optimization steps (the red circle is the optimization level).
![]()
Figure 3. Optimized structure of isopropenyl-4-methyl-1,2-cyclohexanediol and a plot showing its optimization steps (the red circle is the optimization level).
On the other hand, isopropenyl-4-methyl-1,2-cyclohexanediol attained the global minima structure with 51 optimization steps.
4-(2,4-Dimethylcyclohexyl)-2-butanone required fewer optimization steps (36) to attain a structure of minimum energy while the scission of C10, C11 bond occurs with a bond dissociation energy of 494.24 kJ/mol. Clearly, 3,4-dimethyl-3-cyclohexene-1-carbaldehyde, takes the shortest computing time in comparison to 4-(2,4-Dimethylcyclohexyl)-2-butanone, isopropenyl-4-methyl-1,2-cyclohexanediol, and 5-(3-phenylpropanoyl) dihydro-2-(3H)-furanone, respectively.
![]()
Figure 4. Optimized structure of 4-(2,4-dimethylcyclohexyl)-2-butanone and a plot showing its optimization steps (the red circle is the optimization level).
Fundamentally, an optimized structure of a compound or molecule is obtained after a stationary point has been during the optimization steps. At this point, the predicted change in energy is limits to zero and levels off as observed in Figures 1-4. Other parameters that show that optimization of a molecule is complete is the absence of imaginary vibrational frequencies in the output file of the computational platform. In the absence of negative or imaginary frequencies the structure are believed to have reached a global minima and therefore fully optimized. Remarkably, all the structures reported in this work are fully optimized based on the aforesaid arguments.
3.2. Frontier Molecular Orbitals and Electron Density Maps
Conventionally, frontier orbitals are the highest occupied molecular orbitals (HOMO) and the lowest unoccupied molecular orbitals (LUMO). The molecular orbitals and electron density maps are of great significance in understanding the reactivity of molecular species. The HOMO and LUMO orbitals are orbitals that are lie closer in energy of any pair of orbitals given any two molecules which tend to interact more strongly [13]. The HOMO-LUMO band-gap energies for the selected volatile organic compounds considered in this study are shown in Table 1. The reactivity index (HOMO-LUMO gap) of the compounds with small difference implies high reactivity and a large difference implies low reactivity in reactions, therefore as the energy gap between the HOMO and LUMO becomes smaller the rate of reaction is enhanced. 4-(2,4-dimethylcyclohexyl)-2-butanone has the smallest HOMO-LUMO energy gap (0.674 eV) and therefore considered more reactive compared to 3,4-Dimethyl-3-cyclohexene-1-carbaldehyde with a larger band gap of 3.918 eV. 5-(3-phenylpropanoyl)dihydro-2(3H)-furanone and isopropenyl-4-methyl-1,2-cyclohexanediol had band gap energies of 3.393 eV and 1.588 eV, respectively. However, 5-(3-phenylpropanoyl)dihydro-2-(3H)-furanonemay have relatively a large HOMO-LUMO energy gap.
![]()
Table 1. HOMO-LUMO band gap energies for the selected volatile organic compounds.
Evidently, the HOMO-LUMO band-gap for 4-(2,4-dimethylcyclohexyl)-2-butanone is clearly low and therefore could be reactive especially towards biological structures contrary to the fact that ketones are known to be least reactive compared to alcohols and aldehydes. Generally, the band-gap between the HOMO and the LUMO is directly related to the electronic stability of the chemical species [13]. This suggests that 5-(3-phenylpropanoyl)dihydro-2(3H)-furanone having a lower HOMO energy value of −5.560 eV is much more stable making it a good nucleophile compared to 4-(2,4-Dimethylcyclohexyl)-2-butanone which are energetically higher in the LUMO. Figure 5 and Figure 6 report the band gap energies for 4-(2,4-dimethylcyclohexyl)-2-butanone and 5-(3-phenylpropanoyl)dihydro-2-(3H)-furanone, respectively.
The key idea of the frontier molecular orbitals theory rests on the fact that most chemical reactions are dominated by the interaction between these frontier orbitals in an electron donor-acceptor regime. The energy gap between the HOMO-LUMO energy levels is a useful parameter of describing and understanding the reactivity of molecules or compounds.
3.3. Electron Density Contour Maps
Electron density contour maps provide an understanding of the charge distributions on molecules. Electron density maps were done for 3, 4-dimethyl-3-cyclohexene-1-carbaldehyde, 5-(3-phenylpropanoyl)dihydro-2(3H)-furanone, and isopropenyl-4-methyl-1,2-cyclohexanediol as presented in appendices A, B and C. These Figures are critical in determining regions of high electron density within a molecule. Electron distribution gives insight on the behaviour of particular molecules in understanding the nucleophilic and electrophilic sites in molecular structures. Figure 7 and Figure 8 give the 2-D and 3-D electron density maps for 5-(3-phenylpropanoyl)dihydro-2(3H)-furanoneas obtained from Chemissian and Gaussian computational platforms, respectively. Regions of low potential are electron rich compared to other regions in the molecule. Evidently, oxygen atoms in 5-(3-phenylpropanoyl)dihydro-2(3H)-furanone 2-D electron density map has a higher electronegativity value than other atoms (C & H) in the structure. Thus, it has a higher charge density around it compared to other atoms. This is clearly shown by deep red coloration in the 3-D structure (Figure 8).
![]()
Figure 5. The HOMO-LUMO band gap for 4-(2,4-dimethylcyclohexyl)-2-butanone determined using Chemissian.
![]()
Figure 6. The HOMO-LUMO band gap for 5-(3-phenylpropanoyl)dihydro-2-(3H)-furanone determined using Chemissian.
Conservatively, the negative potential sites (red colour) represents regions of electrophilic reactivity and interactions through π-π bonding within the aromatic systems and positive potential sites (green colour) represents regions of nucleophilic reactivity [14] Figure 8, vide supra.
3.4. Mechanistic Degradation of 5-(3-Phenylpropanoyl)Dihydro-2(3H)-Furanone
Mechanistic channels attempt to show how starting materials are converted into products during chemical reactions. Thermal decomposition of fuels leads to emission of numerous particulate matter of various classifications that are of public health interest. These compounds include long chain hydrocarbons,
![]()
Figure 7. 2-D electron density map for 5-(3-Phenylpropanoyl)dihydro-2(3H)-furanone.
![]()
Figure 8. 3-D electron density map for 5-(3-phenylpropanoyl)dihydro-2(3H)-furanoneat an iso-value of 0.020.
oxygenated by-products, aromatic hydrocarbons among other eco-toxic organics [6]. This study has also proposed the mechanistic formation of free radicals from selected oxygenated by-products, which are noted to be precursors of cancer, general respiratory human health and reactive oxidative stress (ROS) well known for their central role in initiating both physiological and pathophysiological signal transduction [15]. Free radicals are generated from both endogenous and exogenous sources and are known to penetrate the body where they are degraded or metabolized and harmful free radicals are generated as by-products [16]. To investigate the energetics of the selected molecular emissions, Gaussian ‘16 software employing the B3LYP correlation function and 6-31G basis sets at DFT level of theory were conducted.
Environmental pollutants blamed for a number of illnesses arise as unwanted by-products in various processes, especially in uncontrolled combustion events such as accidental fires, combustion of transport fuels, cigarette smoking and municipals waste incineration [17]. The major areas of concern focus on the mechanistic formation of environmentally free radicals and their associated impact on human health [18], from chronic respiratory and cardiopulmonary dysfunction as a consequence of oxidative stress induced by reactive oxygen species (ROS) [8].
Clearly, the thermal degradation is predicted to occur via an endothermic energy barriers, Scheme 1 (routes i and iv). Formational of radicals via route (i) proceeds with an energy of 415.71 kJ/mol considered less important to biological systems [19]. The scission of -C-C-bond in route (iv) for the formation of ethylbenzyl radical and 5-oxotetrahydrofuran-2-carbaldehydyl radical occurs with an enthalpy change of 372.31 kJ/mol which is thermodynamically feasible. Interestingly, route (viii) shows a highly exothermic reaction, with bond dissociation energy of −1250.75 kJ/mol. The conversion of free radicals to their corresponding free molecules occurs through exothermic channels, routes (iii) and (v ?ix), scheme 1, vide supra.
Density functional theory calculations have been found to be successful in giving theoretical insights into the reactivity of organic compounds. They provide possible reaction pathways which organic compounds can undergo without necessarily carrying out experimental work which may be both tedious and time consuming. Moreover, computational calculations compliment experimental data. Interestingly, the difference in energy changes between experimental and theoretical work is minimal, and in some cases, computational data has been found to be more accurate than experimental data [20]. Consequently, the energy data reported in this work were derived entirely from computational modeling based on the main objective of this study. There is, however; no known experimental data presented anywhere in literature on this work to necessitate comparative analysis.
4. Conclusion
This study has demonstrated that the thermal decomposition of fuel components of Croton megalocarpus biodiesel proceeds chiefly via endothermic energy barriers to form various radicals which may recombine to form free stable molecules proposed to be toxic to humans and the natural environment. Such toxic molecular products may include ketones aldehydes, benzene and its derivatives, among other harmful compounds. The 5-(3-phenylpropanoyl)dihydro-2(3H)-furanone is more electronegative since it has three oxygen atoms compared to other molecules considered in this study which are more electron withdrawing atoms compared to C and H in the structure. Overall, it can be concluded that 5-(3-Phenylpropanoyl)dihydro-2-(3H)-furanone has more electrophilic reactive sites than 4-(2,4-Dimethylcyclohexyl)-2-butanone, 3,4-dimethyl-3-cyclohexene-1-carbaldehyde and isopropenyl-4-methyl-1,2-cyclohexanediol. This observation is supported by the electron density maps reported in this study.
Acknowledgements
The authors wish to thank the University of KwaZulu-Natal (UKZN), for according JK the opportunity to conduct GC-MS.