A Complete DFT, TD-DFT and Non-Linear Optical Property Study on 6-Amino-2-Methylpyridine-3-Carbonitrile ()
1. Introduction
Since the work reported by O’Regan and Grätzel in 1991 [1] , the metal-free organic sensitizers were investigated as a new generation solar cell of product materials in dye sensitized solar cells (DSSCs), due to their high conversion efficiencies of light-to-electricity, easy structural modifications and cost effective [2] [3] alternatives to conventional photovoltaic materials based on compound semiconductors or inorganic silicon [4] [5] . As a key part of DSSCs, sensibilizers play a role in high power conversion efficiencies (PCEs). Simultaneously, most efficient organic sensibilizers like coumarin [6] [7] , thiophene [8] [9] , indoline [10] [11] , triphenylamine [12] [13] have been tested in this D-π-A framework and good performances is observed. However, considerable progress has been made in the performance of DSSCs by experimental as well as theoretical methods; recently a rapid progress of organic dyes has been witnessed reaching close to 12.5%; that have been reported based on metal-free organic sensitizers [14] . Generally, the factors leading to the low photo-to-current conversion efficiency are the formation of serious intermolecular aggregation on the TiO2 films as well as the charge recombination between the internal and external circuit current.
To have the experimentally observed photophysical properties of design of the 6-Amino-2-methylpyridine-3-carbonitrile dye with the desirable properties, theoretical investigations on the structure property relationship of these materials are essential. Quantum chemical methods play an important role to design and screen out efficient organic dyes reasonably, saving economical cost and synthesis efforts to investigate the relationship between electronic structures and the photophysical properties of the organic molecules [15] [16] [17] . The aim of the present work is to investigate the optimized geometries, polarizabilities and hyperpolarizabilities and the optical absorption properties of 6-Amino-2-me- thylpyridine-3-carbonitriles using DFT and TD-DFT.
In the present study, the optoelectronic properties of the designed molecules were studied using density functional theory (DFT) methods, calculation for the ground state and TD-DFT calculation for the excited state for the various rigid 6-Amino-2-methylpyridine-3-carbonitriles. Based on the optimized geometries we have calculated the absorption and emission spectra using the TD-DFT method with the B3LYP exchange correlation functional using 6-311++G(d,p) basis set. In this work, we computed dipole moment (μ), electronic Polarizability (α), mean first hyperpolarizability (β0) and second order hyperpolarizability (γ).
2. Theory/Computational Method
The geometries of molecule were optimized using the Gaussian 09 suite of programs [18] . The ground state (S0) geometry of the 6-Amino-2-methylpyri- dine-3-carbonitrile dyes were optimized using DFT [19] . The B3LYP method combines Becke’s three parameter exchange functional (B3) [20] with the nonlocal correlation functional by Lee, Yang and Parr (LYP) [21] . The basis set used for all atoms was 6-311++G(d,p). The vertical excitation energies and oscillator strengths were obtained for the lowest twenty singlet-singlet transitions at the optimized the ground state equilibrium geometries by using the Time-Depen- dent DFT (TD-DFT) at the same hybrid functional and basis set [22] [23] . All the calculations were carried out in solvent and gas medium using the Self-Con- sistent Reaction Field (SCRF) under the conductor like Polarizable Continuum Model (C-PCM) [24] [25] . In this work we have also calculated dipole moment (μ), electronic Polarizability (α), mean first Hyperpolarizability (β0) and second Hyperpolarizability (γ) using B3LYP functional with the 6-311++G(d,p) basis set. For completeness, we summarize the essential formulas used in our work, highlighting the quantities in which we are interested. Second-order non-linear optical properties of the 6-Amino-2-methylpyridine-3-carbonitrile chromophore. The components of β0 are defined as the co-efficient in the Taylor series expansion of the energy in the external electric field. When the external electric field is weak and homogeneous, this expansion becomes.
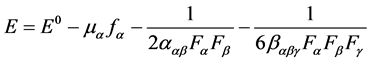 (1)
(1)
where E0 is the energy of the unperturbed molecules, fα is the field at the origin, μα, ααβ and βαβγ are the components of dipole moment, Polarizability and the first Hyperpolarizability, respectively [26] .
The total static dipole moment μ is expressed by following equation
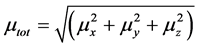 (2)
(2)
The isotropic Polarizability can be calculated from the trace of the polarization tensor,
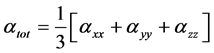 (3)
(3)
Anisotropy of the Polarizability Δα is expressed by
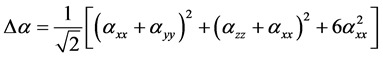 (4)
(4)
The mean first Polarizability (β0) is expressed by
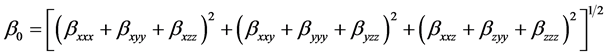 (5)
(5)
where βxxx, βxyy, βxzz, βxxy, βyyy, βyzz, βxxz, βzyy and βzzz are the components of the first order hyperpolarizability tensor along the x, y, and z axes
The mean second Hyperpolarizability (γ) is expressed by
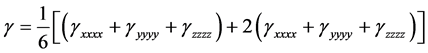 (6)
(6)
3. Results and Discussion
3.1. Geometric Structures in the Ground State
To understand the ground-state geometry of 6-Amino-2-methylpyridine-3- carbonitrile dye have been optimized at B3LYP/6-311++G(d) level of theory using C1 point group. As shown in Figure 1 the bond length C1-C2 is increased from 1.395 to 1.398 Å. This change in the bond length indicates the charge transfer observed from pyridine ring to amino group. There are no changes observed in other bond length like C4-C5, C5-N10. The calculated bond length, bond length and dihedral angle as shown in Table 1.
The dihedral angle C2-C7-N8-C3-C1 for 6-Amino-2-methylpyridine-3-car- bonitrile in acetonitrile solvent in S0 state is 179.89˚ while in S1 state it is 180˚ while this change in the indicating the charge transfer observed in 6-Amino-2- methylpyridine-3-carbonitrile.
![]()
Figure 1. Optimized geometrical structure of dye 6-Amino 2-methylpyridine 3-carboni- trile.
3.2. Molecular Orbital Energies
Energy levels of the frontier molecular orbital’s especially HOMO, LUMO, HOMO-1 and LUMO+1 as well as their spatial distributions are important parameters for determining the optoelectronic properties. The density plot of the HOMO and LUMO of 6-Amino-2-methylpyridine-3-carbonitrile is calculated at B3LYP/6-311++G(d) level of theory and are shown in Figure 2.
The orbital diagrams are plotted with the contour value of 0.02 a.u. The HOMO and LUMO plot of the studied 6-Amino-2-methylpyridine-3-carboni- trile molecules have the typical π molecular orbital characteristics. The HOMO (H)-LUMO (L) energy gap of 6-Amino-2-methylpyridine-3-carbonitriles is found in the range of 5.08 to 4.84 eV in acetonitrile medium. The lowest energy gap is observed for 6-Amino-2-methylpyridine-3-carbonitrile.
3.3. Electronic Absorption Spectra
The calculated vertical excitation spectra associated with their oscillator strength, composition, and assignments of 6-Amino-2-methylpyridine-3-carbonitrile dye in acetonitrile solvent is shown in Table 2. The theoretical λmax reported in the following corresponds to the first singlet-singlet excited states with dipole allowed transition (i.e., oscillator strength f is > 0) from the excited state. The absorption spectra for all the dyes are mainly due to the electronic transition from the HOMO to the LUMO. The calculated absorption wavelength is 398 nm for HOMO → LUMO transition. The absorption spectra for the 6-Amino-2-me- thylpyridine-3-carbonitriles in acetonitrile solvent computed at TD-B3LYP/6- 311++G(d,p) level of theory are shown in Figure 3.
![]()
![]()
Table 1. Bond lengths (in Angstrom) bond angles (in degree) and dihedral angles (in degree) of the dye 6-Amino 2-methylpyridine 3-carbonitrile.
![]()
Table 2. Calculated maximum absorption wavelengths λmax/nm Oscillator strengths (f) and the corresponding electronic transitions assignment for Solvent (Acetonitrile) at TD-B3LYP/6-311++G(dp) level theory of the 6-Amino 2-methylpyridine 3-carbonitrile.
3.4. IR and Raman Frequencies
Figure 4 and Figure 5 show the calculated IR and Raman spectra of 6-Ami- no-2-Methylpyridine 3-carbonitrile. Simulations of calculated IR and Raman spectra have been plotted using pure Lorentzian band shape with a bandwidth (FWHM) of 10 cm−1. These calculations were done by using HF/6˗311++G(d,p) and B3LYP/6˗311++G(d,p). A corrective vibrational scaling factor of 0.9613 [27] [28] for B3LYP calculated frequencies and 0.8982 [29] [30] for HF calculated frequencies were applied to account the anharmonicity. Table 3 presents the main features of HF/6-311++G(d,p) and B3LYP/6-311++G(d,p) calculated vibrational wavenumbers of 6-Amino-2-methylpyridine-3-carbonitrile. FT-IR spectra show six major vibrations viz., 828, 1367, 1647, 2321, 3622 and 3754 cm−1. However, we observed 45 vibrations for 6-Amino-2-methylpyridine-3-carbonitrile but for clarity we consider only major vibrations. The peak at 828 cm−1 corresponds to C-H out of plane bending and 1367, 1647 cm−1 vibration is attributed to (C-N stretching + C-C stretching) and Ring symmetry deformation respectively. The peak observed around 2321 cm−1 can be assigned to C≡N stretching and C-N-N stretching in the nitrile group. The two peaks around 3622 and 3754
![]()
Figure 2. Isodensity plots (isodensity contour = 0 02 a. u) of the frontier orbitals of 6-Amino 2-methylpyridine 3-carbonitrile.
![]()
Figure 3. Experimental and calculated electronic absorption spectra of the dye 6-Amino 2-methylpyridine 3-carbonitrile.
![]()
Figure 4. Observed and calculated FT-IR spectra of 6-Amino 2-me- thylpyridine 3-carbonitrile.
![]()
Figure 5. Observed and calculated FT-Raman spectra of 6-Amino 2- methylpyridine 3-carbonitrile.
cm−1 are attributed to CH-stretching, NH2 asymmetry stretching and NH2 symmetry stretching respectively. We observed six major bands in the Raman spectra of 6-Amino-2-methylpyridine-3-carbonitrile i.e., 1656, 2316, 3033, 3198, 3612 and 3757 cm−1. These bands are almost close observations with IR vibrational bands which confirm that molecule doesn’t show any centro symmetry.
3.5. Nonlinear Optical (NLO) Properties
The computed the static dipole moment (μ), mean Polarizability (α0), polarizability anisotropy (Δα), static first hyperpolarizability (β0) and second hyperpola-
![]()
![]()
Table 3. Comparison of the observed (FT-IR and FT-Raman) and calculated vibrational frequencies of 6-Amino 2-methylpyridine 3-carbonitrile.
rizability (γ) of 6-Amino-2-methylpyridine-3-carbonitrile molecule and which are gathered in Table 4 and Table 5 respectively. As can be seen, the mean polarizabilities of all 6-Amino-2-methylpyridine-3-carbonitrile are almost the same in gas phase as well as in solvent medium. But in case of polarizability anisotropy (Δα), 6-Amino-2-methylpyridine-3-carbonitrile shows higher value (1.322 × 10−24 esu). Such a large value can be attributed to the lower compactness structure of this 6-Amino-2-methylpyridine-3-carbonitrile. The second-order nonlinear optical (NLO) properties originate from the non-centrosymmetric alignment of NLO. The first order Hyperpolarizability of 6-Amino-2-methylpyri- dine-3-carbonitrile calculated at B3LYP/6-311++G(d,p) level. The computed values for the first order Hyperpolarizability (β0) for the 6-Amino-2-methylpy- ridine-3-carbonitrile were found to be greater than urea (3.71028 × 10−31) by 3 times respectively using the B3LYP functional in the Solvent medium. This obtained large β0 value confirms that there should be charge transfer characteristics of the first excited state. The calculated second order hyperpolarizability (γ) is 8.92 × 10−36 e.s.u. and second-order nonlinear optical property is found to be 13 times greater than urea (0.68 × 10−36). This is because the ortho methyl group of the rigid 6-Amino-2-methylpyridine-3-carbonitrile core makes intramolecular hydrogen bonding with nitrogen and makes the structure planar. Due to the planarity, the intramolecular charge transfer (ICT) properties of the 6-Amino-2- methylpyridine-3-carbonitrile dyes get enhanced and result into an increase in β0 value. A molecule with a large dipole moment and high molecular polarizability
![]()
Table 4. The calculated polarizability and Anisotropy of the polarizability components of 6-Amino 2-methylpyridine 3-carbonitrile.
![]()
Table 5. The calculated first order hyperpolarizability components of the 6-Amino 2- methylpyridine 3-carbonitrile.
would be expected to exhibit high photoelectric conversion efficiency. The calculated dipole moment of the 6-Amino-2-methylpyridine-3-carbonitrile molecule charge direction shown in Figure 6. The dipole moment of the dye molecule in the gas phase and solvent medium is 6.06 and 6.41 Debye respectively. From these results, we can say that intramolecular charge transfer would be better facilitated by attachment of an electron-withdrawing amino group.
3.6. Mulliken Population Analysis
The natural population analysis of 6-Amino-2-methylpyridine-3-carbonitrile obtained by Mulliken [31] population analysis with B3LYP using basis set 6-311++G(d,p). The Mulliken charge calculated different levels and at same basis set listed in Table 6. The charge depending on basis set and are changed due to Polarizability. The C9 atom has more negative charge both HF/B3LYP/6- 311++G(d,p), whereas the C2 atom has more positive charge than the other atoms see in Figure 7. The H and C atoms are electron acceptor and the charge transfer takes place from H to C. The N8 and C7 atoms by B3LYP/6-311++G- (d,p) methods are more positive than the other atoms due to electron accepting substitutions at that position in 6-Amino-2-methylpyridine-3-carbonitrile.
4. Conclusion
In the present work, DFT and HF calculations were used to optimize the geometry, structure and electronic properties of 6-Amino-2-methylpyridine-3-carboni- trile dye sensitizer. The strongest IR absorption for 6-Amino-2-methylpyridine- 3-carbonitrile corresponds to the vibrational mode 31 near about 1444 cm−1, which is the C=C stretching bonds. The next stronger IR absorption is attributed to vibrational mode 38 near about 2315 cm−1, corresponding to C-N stretching bonds. The dye has a larger dipole moment in the excited state than in the ground state. The electron absorption spectra lie in visible and UV region. The polarizability and first order hyperpolarizability quantities of the 6-Amino-2- methylpyridine-3-carbonitrile molecule sensitizer are 8.786 × 10−24 and 11.334 × 10−30 respectively. The calculated polarizability anisotropy invariant of 2-Amino- 6-Nitrobenzothiazole is 1.322 × 10−24 e.s.u. In summary, the reported results
![]()
Figure 6. The Dipole moment of the 6-Amino 2-methylpyridine 3-carbonitrile.
![]()
Figure 7. The calculated Mulliken atomic charge distribution of the 6-Amino 2-me- thylpyridine 3-carbonitrile.
illustrate that the 6-Amino-2-methylpyridine-3-carbonitrile molecules have good ability for the NLO properties. The above observations confirm that 6-Amino-2-methylpyridine-3-carbonitrile molecule as a dye sensitizer produces high photo to current conversion efficiency if we use it in practical DSSCs.
![]()
Table 6. The calculated Mulliken atomic charge distribution of the 6-Amino-2-methylpy- ridine-3-carbonitrile.
Acknowledgements
One of the authors M. Prakasam acknowledges Periyar University for financial support in the form of University Research Fellow (URF).