Determination of Inorganic Antimony Species Conversions during Its Speciation Analysis in Soil Using Isotope Dilution Techniques ()
1. Introduction
Antimony and its compounds are considered to be priority pollutants by Environmental Protection Agency of the United States (USEPA) and European Union (EU) [1] Sb toxicity depends on its oxidation state; Sb(III) compounds are ten times as toxic as Sb(V) compounds [2,3]. Therefore, accurate determination of inorganic antimony species distribution in environmental samples is of great importance.
Current measurement procedures like On-line coupling of liquid chromatography with ICP-MS can provide selective, sensitive and rapid measurements for inorganic metallic speciation analysis, but it doesn’t necessarily guarantee accurate analytical results. Independent of the other effects, they are also presented in elemental speciation analysis, varying compositions in chromatographic peaks influence the ionization efficiency in the plasma and, therefore, hinder exact external calibration curve [4] Certified Reference Materials (CRMs) are a useful tool to test for accuracy. Unfortunately, at present they are scarce. Besides, a reliable extraction procedure for a certain material may not be so applied to another material because the adsorption/binding forces of the species of interest to the solid are strongly dependent on the matrix. An alternative approach to the use of CRMs for speciation could be resort to highly qualified primary methods such as Isotope Dilution-Inductively Coupled Plasma Mass Spectrometry (ID-ICP-MS) [5] which provides general accuracy and superior uncertainty over other calibration strategies, including external calibration and standard addition [6]. The high accuracy and precision provided by ID-ICP-MS in the species-specific spiking mode can used to correct for the majority of systematic errors occurred in speciation analysis (non-quantitative separation procedures, signal drifts, quantification measurements errors, etc.). Although isotope dilution ICP-MS has been widely employed for trace element analysis in a wide variety of sample matrixes, its application to species determinations has been limited by the non-availability of commercial species-specific enriched spikes. In species specific mode the original sample is spiked prior to separation with advantage that loss of substance has no effect on the analytical result [7].
However, the sample preparation is the most challengeable step of the analytical speciation procedures preventing that reliable enough speciation results can be achieved routinely. This statement refers specially to the speciation of elements in the solid samples where the major difficulty is not the quantification itself but the quantitative extraction of those compounds from the complex solid sample matrix without degradation or species transformation [8]. Rearrangement reactions between the species and degradation reactions cannot be controlled when more aggressive conditions are used in extraction in the search for quantitative recoveries. In such cases, the speciation information might be altered [9]. It is evident that the border line between quantitative extraction and alteration of the species is probably small and will depend on the matrix itself and on the labiality of the sought species [10].
Speciated isotope dilution mass spectrometry (SIDMS) takes a unique approach to speciated analysis that differs from traditional methods. Traditional speciation methods inherently measure the species after species conversion has occurred. SIDMS has been developed to address the correction for the species conversion. In SIDMS, each species is labeled with a different isotope-enriched spike in the corresponding species form. Thus, the interconversion that occur after spiking is traceable and can be corrected. While SIDMS maintains the advantages of IDMS, it is capable of correcting the degradation of the species or the interconversion between the species [11]. Evaluating and correcting for these species transformations in the sample preparation step could be attained if a spike solution containing inorganic antimony species isotopically labeled with different antimony isotopes. This can not be done in a similar way to previous studies which applied SIDMS successfully for the elements having more than two naturally isotopes like Cr [12], since Sb has only two isotopes. For this purpose a special approach is presented in this work. The Sb species distribution after the chromatographic separation was determined using on-line isotope dilution [13] after their extraction from a contaminated soil sample using 100 mmol·L−1 citric acid. The Sb(III) and Sb(V) concentrations at each investigated step in the analytical procedure were determined using species specific spikes [121Sb(III) and 123Sb(V)]. The influence of each step on species transformation can be estimated by subtracting the final concentration of each species done by on-line ID from its concentration done by species specific ID at the investigated step of analytical procedure.
2. Experimental
2.1. Sample
The soil sample examined in this work consisted of 20 subsamples and represented a road section between 50 - 100 m. The subsamples were taken from the soil surface (0 - 5 cm) near the edge of a highway (Vienna/A23) [14]. The collected soil sample was dried at ambient air temperature and then sieved to obtain the <2 mm soil fraction. The sieved sample was grinding using a mixer mill. The final fineness of samples particles after grinding was about 0.01 mm. The sample is an alkaline soil (pH of water extract (1:10) is 8.6) and the organic carbon was 1.71% m·m−1. The soil sample contains 4.17 mg·g−1 as total Sb [13].
2.2. Instrumentation for Hyphenation
A standard Agilent Technologies HP4500 quadruple inductively coupled plasma mass spectrometry (QICP-MS) system was used with Babington for Sb speciation and a cooled quartz glass spray chamber was used. For the chromatographic module a quaternary HP 1050 series pump was used. Separation of Sb(III) and Sb(V) was carried out on a Hamilton PRP-X100 anion exchange column (150 mm × 4.6 mm i.d.) using 10 mmol·L−1 EDTA, 1 mmol·L−1 phthalic acid at pH 4.5 as a mobile phase of a f1ow rate of mL·min−1. Samples and standards were injected using an autosampler (HP 1050 Series) with a l00 µL injection loop. The chromatographic system was interfaced with an ICP-MS instrument using 250 mm of PEEK tubing (0.54 mm i.d.) to connect the column to the nebulizer. On-line isotope dilution was preformed by a continuous addition (0.5 mL of min−1) of the enriched spike was achieved by joining the spike solution line, pumped by a peristaltic pump equipped with a silicone tube (1.02 mm i.d.), with the HPLC effluent (1 mL·min−1) via a three-way valve. The mixed solution passed directly to the nebulizer of the ICP-MS. The “time-resolved analysis” mode of the ICP-MS software was used for the data acquisition. The integration times of Sb isotopes were 0.9 s per mass.
2.3. Chemicals and Reagents
A Milli-Q (Millipore) purified water (18.2 MW·cm−1) was used for the preparation of all solutions. Extraction solutions were prepared from citric acid monohydrate (99.5% - 100.5% mm−1, Merck). The HPLC eluents were prepared with adequate concentrations by dissolving appropriate amounts phthalic acid (≥99.5% mm−1, Fluka) and EDTA disodium salt (99.0% - 101.0% mm−1, Baker). The pHs of these eluents were adjusted by adding ammonia solution (25%, Merck) or nitric acid (200 mmol·L−1). The resulting solutions were de-gassed before using. natSb(V) standard solution with natural isotopic abundance was prepared from antimony(V) oxide (99.995% mm−1, Aldrich). Sb(V) stock solution (1 g » 10 mg Sb) enriched with (94.2%) in 123Sb was obtained as antimony oxide (Sb2O5) [lot number H3451] from Euriso-top France. Procedure describes antimony oxide as source material for 123Sb(V) and natSb(V) illustrated elsewhere [13]. natSb(III) stock solution, 1 g = 0.5 mg Sb was prepared by dissolving 50 mg of Sb metal in 5 mL boiled concentric HCl. Then 2 mL of a reducing solution (40% KI + 5% ascorbic acid) were added to reduce all Sb to Sb(III). The solution was diluted to 100 g using highly purified water. The enriched isotope metal 121Sb(III) (98.73% of 121) has been obtained from Euriso-top (France) [lot number H2841]. It was prepared in the same manner as the natural Sb(III). Reverse isotope dilution was used to find out the exact concentration of 121Sb(III) by calibrating with the natSb(III) solution and independently, with the certified 10 mg·mL−1 ICP-MS standard solution after reduction of possible Sb(V) species using the reducing solution mentioned above. All standard and stock species solution were stored in dark polymeric containers in a refrigerator at a temperature 1˚C - 4˚C.
2.4. Analytical Procedures
2.4.1. Species Unspecific Isotope Dilution
100 μL of extraction solution was injected in the HPLC using the autosampler. From the measured intensities of the corresponding isotopes 121Sb and 123Sb the isotope ratio R123/121(t) for every measured time was calculated. The isotope ratio chromatograms were converted into mass flow chromatograms by calculating the absolute Sb amount for each R123/121(t) on the chromatogram by applying on-line isotope described on our previous work [13]. The background correction was done by subtracting the mean values of Sb mass before and after each peak from each point of the peaks in the mass chromatogram. The exact concentrations of Sb(III) and Sb(V) were determined by integration of the areas under the peaks after background subtraction. A special protocol for HPLCID-ICP-MS measurement (Figure 1) was followed in order to correct for mass bias which is the deviation of the measured ratio of a natural Sb solution from the agreed isotope ratio (123/121Sb 0.74785) [15] and to assume the blank contribution to the R123/121(t) of each measurement.
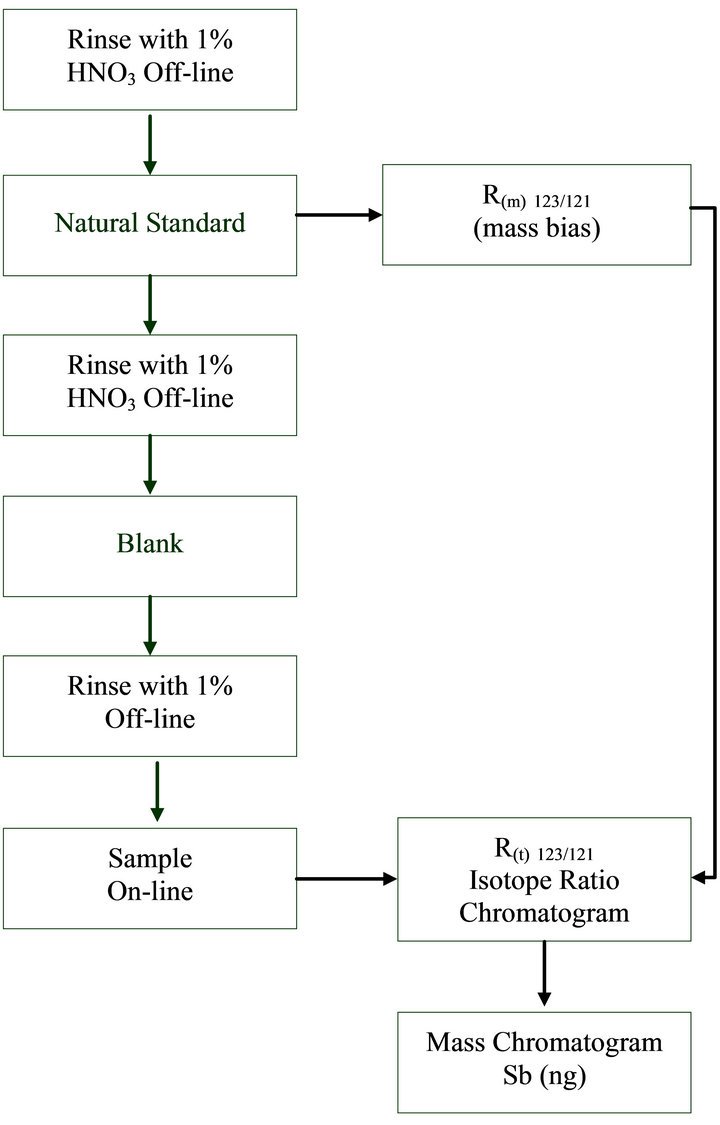
Figure 1. Protocol for HPLC-ID-ICP-MS measurements.
2.4.2. Species-Specific Isotope Dilution
Species-specific isotope dilution procedure which was used to determine the concentrations of inorganic Sb separately in two stages of the speciation analysis procedure is explained below:
1) Before extraction step 500 mg of a representative soil sample were weighed to the nearest 0.01 mg into a 10 mL screwed-cap polystyrene tube. This sample was spiked with 1.00 g of a calibrated isotope-enriched species 121Sb(III) 0.93 mg·g−1 or 123Sb(V) 2.91 mg·g−1 to quantify Sb(III) and Sb(V) at the spiking stage separately. Then soil and the spike solution were shaken manually and the suspension was left for 10 min in a refrigerator at less than 4˚C. The spike species and the original species were extracted using 10 ml of 100 mmol·L−1 citric acid solution using an ultrasonic bath (power 120 W) for 45 min where equilibration can be achieved. The solution was then centrifuged at a speed of 4000 rpm for 5 min. and filtered using a 0.45 μm syringe filter. The filtrate was stored in polystyrene vials in a refrigerator at a temperature ≤ 4˚C until the analysis was possible. Before analysis 3.0 g of spiked soil extract was diluted to 5.0 g using Milli-Q water. Then 100 μL of the diluted extraction solution was injected in the HPLC using autosampler. From the measured intensities of the corresponding isotopes 121Sb and 123Sb the isotope ratios R121/123 for Sb(III) and R123/121 for Sb(V) were calculated from the measured of the peak area ratios after mass bias corrections. In order to correct for mass bias a soil extract obtained in the same way as the spiked soil portion was used before each measurement where the soil extract was assumed to have a natural Sb isotopic composition. The corrected isotope ratios Sb(III)121/123 calculated from the chromatogram obtained for spiked soil sample with enriched 121Sb(III) were used to calculate Sb(III) concentration at this step. While Sb(V) concentration was calculated using the corrected isotope ratio of Sb(V)123/121 obtained from another portion of sample spiked with 123Sb(V). Calculations for element species concentrations are illustrated elsewhere [16].
2) After extraction and before HPLC separation Soil extraction was carried out in the same way as mentioned above for the spiked sample portions. 3.0 g of soil extract was spiked with 1.0 g of calibrated isotopeenriched species 121Sb(III) 0.93 mg·g−1 or 1.0 g of 123Sb(V) 2.91 mg·g−1 to quantify Sb(III) and Sb(V) and the spiked soil extract was diluted with highly purified water to 5.0 g. Then the determinations of species were done in a similar way to that used in case of the spiked soil portions (described above).
3. Results and Discussion
3.1. Interconversion of Inorganic Antimony Species during Its Speciation Analysis in Soil
3.1.1. Purity of 121Sb(III) and 123Sb(V) Spikes
Figure 2(a) represents a diluted solution (1:10) of 121Sb(III) standard after the separation and determination using HPLC-ICP-MS. This solution was used for spiking of the soil portions and soil extracts. It is clear that only Sb(III) enriched with 121Sb is found in this spike solution and no Sb(V). This is very important hence the presence of species specific spike is one of the most important steps in the methodology of species specific isotope dilution quantification. The source (metal) for this spike solution preparation has a certified enriched ratio of the isotope 121% of 98.73%. While this ratio was changed to 81.18% during the spike solution preparation and in the separation step where a lot of chemicals had been used: acids, reducing agents, mobile phase furthermore the complexing reagent. Thus it is important that the exact concentration and enriched isotope percent must be calibrated before spiking the sample. This was done using reverse isotope dilution. This diluted solution was spiked with a known amount of a natSb(III) species of known concentration. The original source which was used to prepare the enriched isotope species 123Sb(V) was Sb2O5 where Sb is enriched with 123 isotope with a percent of 94.2%. The fraction of 123Sb in Sb(V) species decreased after its preparation and separation to 74.05% Figure 2(b). The exact concentration of the 123Sb(V) spike was determined using reverse isotope dilution.