 Journal of Cancer Therapy, 2013, 4, 32-40 http://dx.doi.org/10.4236/jct.2013.47A006 Published Online August 2013 (http://www.scirp.org/journal/jct) Exploiting MCF-7 Cells’ Calcium Dependence with Interlaced Therapy Jonathan Pottle1, Chengrong Sun1, Lloyd Gray2, Ming Li1* 1Department of Physiology, School of Medicine, Tulane University, New Orleans, USA; 2Tau Therapeutics, LLC, Charlottesville, USA. Email: *mli@tulane.edu Received May 3rd, 2013; revised June 4th, 2013; accepted June 12th, 2013 Copyright © 2013 Jonathan Pottle et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. ABSTRACT The purpose of this study is to demonstrate MCF-7 cells’ dependence on calcium for growth and to exploit that de- pendence to improve chemotherapy efficacy. Fura-2 fluorescence imaging shows that MCF-7 cells maintain a higher basal intracellular calcium concentration than non-tumorigenic MCF-10A cells. Blocking T-type calcium channels with mibefradil reduced MCF-7 intracellular calcium concentration. Flow cytometry shows that knocking down T-type cal- cium channel expression with siRNA caused an increase in MCF-7 cells in G1 phase and a decrease in cells in S phase. Proliferation assays of MCF-7 cells treated with EGTA and thapsigargin reveal the dependence of MCF-7 cell growth on extracellular and intracellular calcium sources, respectively. In vitro, interlaced treatment that alternated the T-type calcium channel blocker NNC-55-0396 with paclitaxel more effectively reduced MCF-7 cell number than chemother- apy alone. In a mouse in vivo model, interlaced mibefradil and paclitaxel more effectively reduced MCF-7 xenograft size than chemotherapy alone. These findings indicate that MCF-7 cells are dependent on calcium for proliferation, par- ticularly in passing the G1/S cell cycle checkpoint. Further, this dependence on calcium can be exploited by alternating treatment with T-type calcium channel blockers with paclitaxel in an interlaced therapy scheme that increases the effi- cacy of the chemotherapy. Keywords: Breast Cancer; Calcium; Calcium Channel Blockers; Ion Channels; Interlaced Therapy 1. Introduction Any given cell type expresses a unique cadre of calcium signaling molecules that allows that cell to generate and respond to various calcium signals [1]. The many roles of calcium in eukaryote cell biology are so wide ranging in type and scope that the complement of calcium signaling modalities in a cell must be subject to exquisite regula- tion in order to maintain normal growth and function. Permanent alteration of a calcium signaling mechanism can have a profound impact on the biology of a cell. The abnormal growth characteristic to cancer is often associ- ated with dysfunctional calcium signaling. Elevated cal- cium levels normally cause keratinocytes to differentiate, but transformed keratinocytes are far less likely to un- dergo differentiation regardless of calcium concentration [2]. The calcium binding protein S100-A8, which con- tains two EF-hand motifs and normally participates in cell cycle regulation and differentiation, has been shown to influence the activity of telomerase [3], an enzyme that lengthens telomeres and allows for unlimited chro- mosomal replication in cancer cells. Expression and function of the sarco/endoplasmic reticulum calcium ATPase, a pump that stores calcium ions in the endo- plasmic reticulum lumen, is important for proliferation in LNCaP prostate cancer cells [4]. Calcium channels, which conduct calcium ions from the extracellular envi- ronment or intracellular stores into the cytosol, have been found to play roles in cancer cell proliferation. A mem- ber of the transient receptor potential superfamily of ion channels, TRPV6, has been shown to be essential to pro- liferation in some prostate cancer cells [5]. Voltage-gated T-type calcium channels, characterized by a low thresh- old for activation, have been found to be broadly ex- pressed in cancer cells [6], and their role in proliferation is well observed in cancer cells from a wide variety of tissues of origin. MCF-7 breast cancer cells express T-type calcium channels that conduct current and play a functional role in proliferation [7]. In particular, blocking T-type calcium channels with pharmacological agents and knocking down channel expression with RNA inter- *Corresponding author. Copyright © 2013 SciRes. JCT  Exploiting MCF-7 Cells’ Calcium Dependence with Interlaced Therapy 33 ference reduced the proliferation of MCF-7 cells. Similar findings indicating a dependence of cancer cell prolifera- tion on T-type calcium channels have been reported in esophageal cancer cells [8], retinoblastoma cells [9], co- lorectal and gastric cancer cells [10], and ovarian cancer cells [11] that express T-type calcium channels. Overex- pression of T-type calcium channels doubled the prolif- eration rate of astrocytoma and neuroblastoma cells that already expressed the channels, while antisense oligonu- cleotides directed against mRNA coding for T-type cal- cium channels reduced the proliferation rate in these cells but had no effect in cancer cells that lack T-type calcium channel expression [12]. Similar results were found the prostate cancer cell line LNCaP, in which T-type calcium channels play a role in intracellular calcium regulation [13,14]. In this study, we set out to investigate the physiology of calcium in breast cancer cells by estimating the intra- cellular calcium concentration in cancerous and non- cancerous cell lines, and by assessing the importance of extracellular and intracellular calcium sources to breast cancer cell growth. We also examined how T-type cal- cium channel inhibition can affect passage through the cell cycle, and we employed both in vitro and in vivo models to determine if these channels may be potential targets in breast cancer therapy. 2. Materials and Methods 2.1. Cell Culture MCF-7 cells were cultured in Dulbecco’s Modified Ea- gle’s Medium (DMEM; ATCC, Manassas, VA) supple- mented with 10% fetal bovine serum (FBS; Gibco by Life Technologies, Carlsbad, CA), 1% pen/strep (Gibco), and 0.01 mg/mL insulin (Gibco). Non-tumorignenic MCF-10A cells were grown in serum-free Mammary Epithelial Growth Medium (MEGM; Lonza Group Ltd., Basel, Switzerland) supplemented with 100 ng/mL chol- era toxin. Culture flasks, plates and dishes were incu- bated in a humidified, 37˚C incubator. For subculturing, growth medium was removed by vacuum and the cell monolayer was rinsed with DPBS (Gibco). The mono- layer was detached by addition of trypsin/EDTA (ATCC) followed by incubation for 5 minutes. For MCF-7 cells, complete growth medium was added to neutralize the trypsin, and the cell suspension was centrifuged at 1500 rpm. In the culture of MCF-10A cells, to neutralize tryp- sin soybean trypsin inhibitor (0.5 mg/mL; Gibco) was added in volume equal to that of the trypsin solution. Cells were resuspended in complete growth medium and split at 1:3 - 1:6. 2.2. Fura-2 Fluorescence Intracellular calcium concentrations were estimated with the calcium-binding fluorophore fura-2 AM (Molecular Probes by Life Technologies). Cells were plated on poly-D-lysine-coated glass coverslips and cultured over- night. The blank measurement solution contained, in mM: 130 NaCl, 5 KCl, 1 MgCl2, 2 CaCl2, 15 HEPES; solution pH was adjusted to 7.4 with NaOH. Cells were loaded with the fura-2 AM loading solution, consisting of meas- urement solution plus 3 - 5 μM fura-2 AM (stock in DMSO; 3 - 5 μL stock per mL of loading solution) and an identical volume of a 10% pluronic acid solution (Sigma-Aldrich), in a humidified 37˚C incubator for 1 h. Cells were then washed thrice with measurement solution, then incubated in measurement solution for 30 minutes to allow for fura-2 AM deesterification. Coverslips bearing fura-2 AM loaded cells were mounted in an Attofluor cell chamber (Molecular Probes) for analysis. Excitation wavelengths were 340 nm and 380 nm, and emission was measured at 510 nm. The emission intensities measured for 340 nm and 380 nm were used to calculate an emis- sion intensity ratio, which can be converted to free cal- cium concentration. Fluorescence measurements were taken with an inverted microscope in conjunction with a 175 watt xenon arc lamp and Metafluor imaging system (Nikon Instruments, Melville, NY). The fluorescent emis- sion signal was captured by an ADC 20 MHz Photo- metric Coolsnap FX monochrome camera (Nikon). Data were analyzed with Metafluor software (Molecular De- vices, LLC, Sunnyvale, CA). To measure basal calcium, MCF-7 or MCF-10A cells were subjected to ratiometric fluorescent analysis with blank measurement solution in the cell chamber. To test the effects of inhibiting T-type calcium channel window current on the basal calcium concentration in MCF-7 cells, measurement solution with 3 mΜ mibefradil (Sig- ma-Aldrich) was perfused in the cell chamber. Blank measurement solution was perfused through the chamber to serve as a perfusion control. To mitigate calcium buff- ering from intracellular stores in the mibefradil experi- ments, measurement solution with 1 μM thapsigargin (an irreversible inhibitor of the sarcoplasmic/endoplasmic reticulum calcium ATPase; Sigma-Aldrich) was perfused through the chambde hydrostatic pressure driving force. Solution flow rates were approximer prior to addition of mibefradil. Solution perfusion was controlled by a War- ner Instruments valve controller (Hamden, CT), with solution reservoirs elevated to proviately 5 mL/min. Sin- gle-cell epifluorescence measurement images were cap- tured throughout the experiments. Exposure times were initially adjusted to maximize usage of the camera’s dy- namic range, but kept constant throughout a given ex- periment. 2.3. Proliferation Assay To assess the effects of a particular agent on the prolif- Copyright © 2013 SciRes. JCT  Exploiting MCF-7 Cells’ Calcium Dependence with Interlaced Therapy 34 eration/survival of MCF-7 cells in vitro, proliferation assays were performed. Cells were plated in 96 well plates and cultured overnight in a humidified, 37˚C in- cubator. The next day, experimental medium was pre- pared by mixing drug stocks into complete growth me- dium to achieve the desired concentration, with stock solvent added to make vehicle concentrations equal across all concentrations of the agent of interest. EGTA was dissolved in distilled water to 10 mM for working stock, then diluted in complete medium to concentrations of (in μM): 1.0, 1.5, 2.0, and 3.0. Thapsigargin was dis- solved in DMSO to 1 mM for working stock and diluted in complete medium to experimental concentrations (in nM): 10, 30, 100, 300, and 1000. Mibefradil and NNC- 55-0396 were dissolved at 30 mM in distilled water, then diluted to 3 mM for working stock; experimental con- centration was 10 μM in complete medium. Paclitaxel was dissolved in DMSO to 1 mM, and diluted in DMSO to 100 μM working stock; experimental concentration was 10 nM. For vehicle control medium, only stock sol- vent was added to complete growth medium. The culture medium was removed from plate wells and 0.1 mL of the appropriate experimental medium was added to each well for the appropriate experimental time period. Cell num- ber was WST-8 assay kit (Cell Counting Kit-8; Dojindo Molecular Technologies, Kumamoto, Japan) according to the manufacturer’s instructions. Absorbance at 540 nm was measured with a Multiskan Multisoft multi-well rea- der. 2.4. RNA Interference T-type calcium channel expression levels were knocked down with short interfering RNA (siRNAs) designed to target both the α1G and α1H isoforms of T-type calcium channels, as described earlier [7]. The sequence targeted by the siRNAs used were: scrambled control 5’-UAGUGAAGGGAGUCGGAUCUC-3’, T-type 5’-GCCAUCUUCCAGGUCAUCACA-3’. MCF-7 cells were plated in 96 well plates and cultured overnight in a humidified 37˚C incubator. Cells were transfected with 100 nM siRNA, aided by Lipofectamine 2000 (Invitro- gen) according to the manufacturer’s instructions. Re- duced mRNA levels were verified with quantitative RT- PCR (data not shown). 2.5. Flow Cytometry To assess the effects of inhibiting T-type calcium chan- nels on passage of MCF-7 cells through the phases of the cell cycle, flow cytometry experiments were performed. MCF-7 cells were plated in culture dishes and cultured overnight. Experimental treatments were applied the fol- lowing day and maintained for the appropriate lengths of time. At the end of the experimental period, cells were collected and suspended in 5 mL PBS, then centrifuged. After resuspension in 0.5 mL PBS, the cells were added to 4.5 mL 70% ethanol for fixing overnight. The next day, the fixed cells were centrifuged and the ethanol solution decanted. The cell pellet was resuspended in 10 mL PBS with 0.1% v/v Triton X-100 (Sigma-Aldrich), 2 mg DNAse-free RNAse (Sigma-Aldrich) and 200 μL of 1 mg/mL propidium iodide (PI), and the suspension was incubated at room temperature for 30 min. A BD LSR II (BD Biosciences, San Jose, CA) flow cytometer was used to measure cell fluorescence in flow cytometry, with excitiation with blue light and detection of PI emission with red light. The DNA content frequency histogram was analyzed for cell-cycle progression us- ing ModFit LT v3.0 software (Verity Software House, Maine). 2.6. In Vivo Xenografts Ovariectomized 25 - 35 days old nu/nu athymic female mice (Charles River Laboratories, Wilmington, MA) were obtained and allowed to acclimate to the vivarium environment for 1 week. Mice then had slow-release 17-beta-estradiol pellets (0.72 mg/60 days relase) im- planted subcutaneously with a trochar. MCF-7 cells were cultured in standard conditions in 225 cm2 cell culture flasks, harvested via trypsinization, and 5 * 106 cells (in 80 μL PBS) were mixed with cold Matrigel and kept on ice to maintain the fluidity of the gel. Aliquots of the Matrigel-cell suspension (80 μL, BD Biosciences) were implanted in the abdominal mammary fat pads of the mice with surgical needle-syringes to form tumor xeno- grafts. All implantations followed anesthetization with ketamine/xylazine (0.88 ml/kg, intraperitoneal). After 5 days for tumor establishment, mice were randomized into four treatment groups: control (estradiol pellets only), mibefradil only, paclitaxel only, and alternating mibe- fradil/paclitaxel. Mibefradil (Sigma-Aldrich) was sus- pended in distilled water (6 mg/mL) and administered by oral gavage at 30 mg/kg for 3 days every week. Pacli- taxel (Sigma-Aldrich) stock, 32 mg/mL, was made in ethanol/Cremphor EL (polyethoxylated castor oil, glyc- erol polyethelene glycol ricinoleate) solution. For ad- ministration, paclitaxel stock was diluted in PBS to 2.4 mg/mL and administered via IP injection once per week (12 mg/kg in 0.1 mL). The control group received no treatment, the mibefradil group received mibefradil treatment via oral gavage only, the paclitaxel group re- ceived paclitaxel treatment via IP injection only, and the alternating group received both treatments. Tumor dimensions were monitored with digital cali- pers thrice weekly, with volumes calculated by the for- mula Volume = 0.5 * width2 * length. Mice body weights were also measured thrice weekly, as a general measure of animal health. At completion of the experiment (35 days), the mice were sacrificed and tumors, hearts, livers, Copyright © 2013 SciRes. JCT 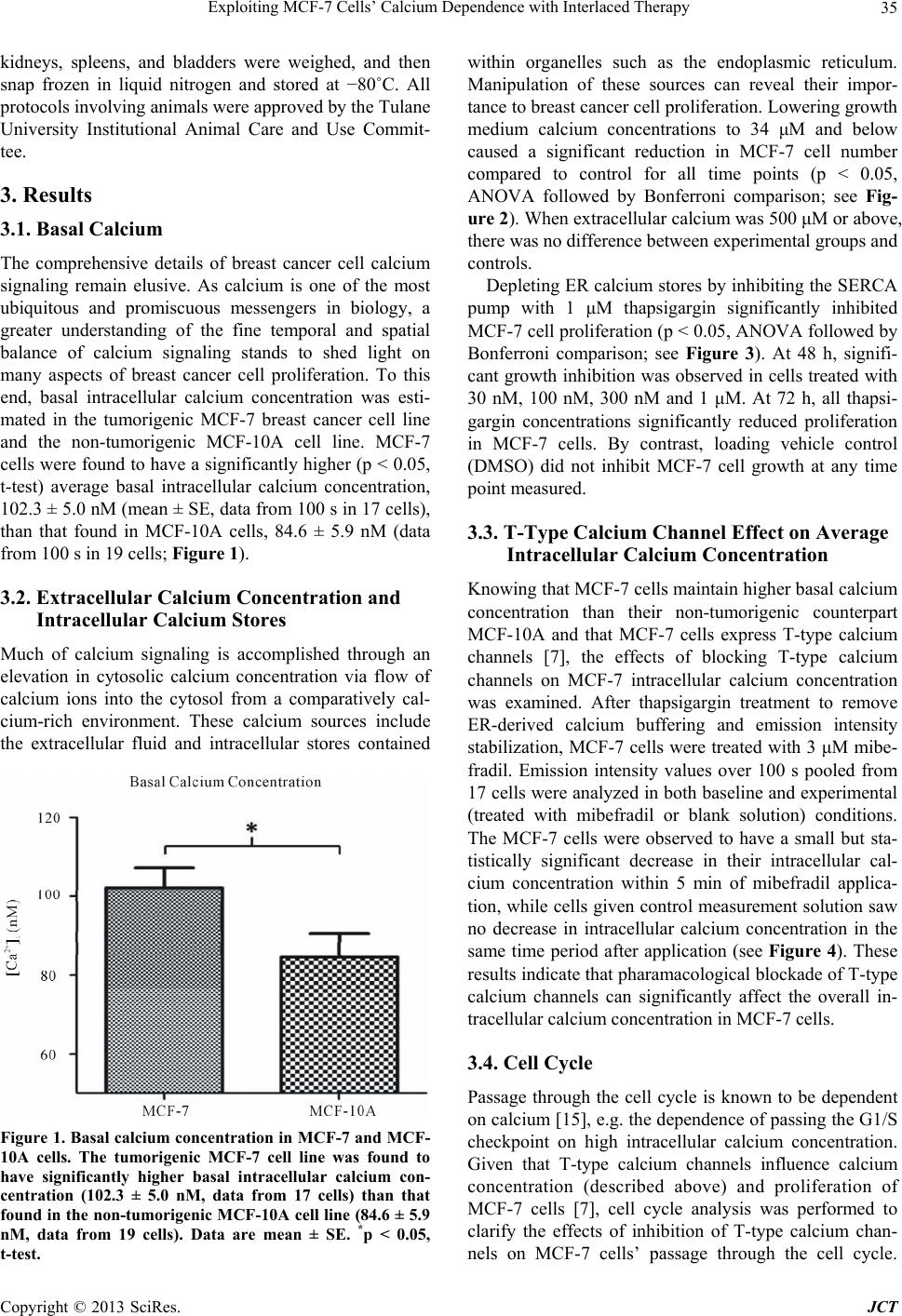 Exploiting MCF-7 Cells’ Calcium Dependence with Interlaced Therapy 35 kidneys, spleens, and bladders were weighed, and then snap frozen in liquid nitrogen and stored at −80˚C. All protocols involving animals were approved by the Tulane University Institutional Animal Care and Use Commit- tee. 3. Results 3.1. Basal Calcium The comprehensive details of breast cancer cell calcium signaling remain elusive. As calcium is one of the most ubiquitous and promiscuous messengers in biology, a greater understanding of the fine temporal and spatial balance of calcium signaling stands to shed light on many aspects of breast cancer cell proliferation. To this end, basal intracellular calcium concentration was esti- mated in the tumorigenic MCF-7 breast cancer cell line and the non-tumorigenic MCF-10A cell line. MCF-7 cells were found to have a significantly higher (p < 0.05, t-test) average basal intracellular calcium concentration, 102.3 ± 5.0 nM (mean ± SE, data from 100 s in 17 cells), than that found in MCF-10A cells, 84.6 ± 5.9 nM (data from 100 s in 19 cells; Figure 1). 3.2. Extracellular Calcium Concentration and Intracellular Calcium Stores Much of calcium signaling is accomplished through an elevation in cytosolic calcium concentration via flow of calcium ions into the cytosol from a comparatively cal- cium-rich environment. These calcium sources include the extracellular fluid and intracellular stores contained Figure 1. Basal calcium concentration in MCF-7 and MCF- 10A cells. The tumorigenic MCF-7 cell line was found to have significantly higher basal intracellular calcium con- centration (102.3 ± 5.0 nM, data from 17 cells) than that found in the non-tumorigenic MCF-10A cell line (84.6 ± 5.9 nM, data from 19 cells). Data are mean ± SE. *p < 0.05, t-test. within organelles such as the endoplasmic reticulum. Manipulation of these sources can reveal their impor- tance to breast cancer cell proliferation. Lowering growth medium calcium concentrations to 34 μM and below caused a significant reduction in MCF-7 cell number compared to control for all time points (p < 0.05, ANOVA followed by Bonferroni comparison; see Fig- ure 2). When extracellular calcium was 500 μM or above, there was no difference between experimental groups and controls. Depleting ER calcium stores by inhibiting the SERCA pump with 1 μM thapsigargin significantly inhibited MCF-7 cell proliferation (p < 0.05, ANOVA followed by Bonferroni comparison; see Figure 3). At 48 h, signifi- cant growth inhibition was observed in cells treated with 30 nM, 100 nM, 300 nM and 1 μM. At 72 h, all thapsi- gargin concentrations significantly reduced proliferation in MCF-7 cells. By contrast, loading vehicle control (DMSO) did not inhibit MCF-7 cell growth at any time point measured. 3.3. T-Type Calcium Channel Effect on Average Intracellular Calcium Concentration Knowing that MCF-7 cells maintain higher basal calcium concentration than their non-tumorigenic counterpart MCF-10A and that MCF-7 cells express T-type calcium channels [7], the effects of blocking T-type calcium channels on MCF-7 intracellular calcium concentration was examined. After thapsigargin treatment to remove ER-derived calcium buffering and emission intensity stabilization, MCF-7 cells were treated with 3 μM mibe- fradil. Emission intensity values over 100 s pooled from 17 cells were analyzed in both baseline and experimental (treated with mibefradil or blank solution) conditions. The MCF-7 cells were observed to have a small but sta- tistically significant decrease in their intracellular cal- cium concentration within 5 min of mibefradil applica- tion, while cells given control measurement solution saw no decrease in intracellular calcium concentration in the same time period after application (see Figure 4). These results indicate that pharamacological blockade of T-type calcium channels can significantly affect the overall in- tracellular calcium concentration in MCF-7 cells. 3.4. Cell Cycle Passage through the cell cycle is known to be dependent on calcium [15], e.g. the dependence of passing the G1/S checkpoint on high intracellular calcium concentration. Given that T-type calcium channels influence calcium concentration (described above) and proliferation of MCF-7 cells [7], cell cycle analysis was performed to clarify the effects of inhibition of T-type calcium chan- nels on MCF-7 cells’ passage through the cell cycle. Copyright © 2013 SciRes. JCT  Exploiting MCF-7 Cells’ Calcium Dependence with Interlaced Therapy Copyright © 2013 SciRes. JCT 36 Figure 2. Effects of EGTA on MCF-7 cell proliferation. EGTA, a calcium chelator, has a significant inhibitory effect on MCF-7 cell proliferation compared to control at concentrations equal to or greater than 2 mM (corresponds to free calcium concentration of 34 μM or lower), p < 0.05 for 2 mM and 3 mM EGTA. A higher concentration of EGTA in the growth me- dium results in a lower extracellular calcium concentration. Data normalized to that of cells in untreated control medium. n = 6. Data analyzed with ANOVA for overall difference, then columns compared to medium control with Dunnett’s multiple comparison test. Figure 3. Effects of thapsigargin on MCF-7 cell proliferation. Thapsigargin, an inhibitor of the sarcoplasmic/endoplasmic calcium ATPase, exerts time-dependent and dose-dependent inhibitory effect on MCF-7 cell proliferation. Data normalized to that of cells in untreated control medium. n = 6. Data analyzed with ANOVA for overall difference, then columns compared to medium control with Dunnett’s multiple comparison test.  Exploiting MCF-7 Cells’ Calcium Dependence with Interlaced Therapy 37 Figure 4. MCF-7 intracellular calcium concentration in response to treatment with mibefradil. Fura-2 fluorescence shows that blockade of T-type calcium channels with 3 μM mibefradil results in a significant decrease compared to basal level in MCF-7 intracellular calcium concentration within 120 s of mibefradil application. The intracellular calcium concentration of MCF-7 cells treated with control solution (Blank) maintained basal level with no significant change. Data from 100 s for each condition, pooled from 17 cells. Data analyzed with t-test, *p < 0.05 vs. baseline. MCF-7 cells were treated with siRNA targeted against T-type calcium channel mRNA (siRNA-2, see above) or scrambled siRNA, and then subjected to flow cytome- tryto assess their DNA content and thus infer their cell cycle stage. Treatment with siRNA targeted against T- type calcium channel mRNA resulted in a large increase in the percentage of MCF-7 cells in G1 phase and a smaller increase in cells in G2, along with a decrease in cells in S phase, compared to cells treated with scram- bled siRNA (Figure 5 and Table 1). This indicates that functional inhibition of T-type calcium channels reduces proliferation largely through halting cells in the G1 phase, preventing them from passing through the checkpoint into S phase. 3.5. Interlaced: In Vitro Since T-type calcium channels play a functional role in MCF-7 cell growth [7], T-type calcium channels may be a viable target in breast cancer treatment. By virtue of their role in the cell cycle, blocking T-type calcium channels may serve as a means of reducing tumor cell growth in between applications of cytotoxic chemotherapy. To as- sess this possibility, MCF-7 cells were cultured in 96-well plates and subjected to a scheme that interlaced 3 μM NNC-55-0396 with 10 nM paclitaxel, each drug ap- plied for 24 h. While treatment with NNC-55-0396 ini- tially was not as effective as treatment with paclitaxel, by the end of the experiment at 72 h, the cells receiving in- terlaced NNC-55-0396 and paclitaxel were significantly reduced in number compared to cells receiving either Figure 5. Flow cytometry of MCF-7 cells treated with siRNA directed against T-type calcium channel mRNA. RNA in- terference directed against T-type calcium channel mRNA causes a marked decrease in the proportion of MCF-7 cells in S-phase, with most cells shifted into G1, compared to cells treated with scrambled siRNA. Table 1. Cell cycle analysis of MCF-7 cells treated with siRNA-T. Total G1 % Total S % Total G2 % siRNA-T 70.33 11.85 17.82 Scrambled 61.07 25.13 13.80 Cell cycle analysis of MCF-7 cells treated with siRNA targeting T-type calcium channel mRNA (siRNA-T) compared with cells treated with control (scrambled) siRNA. treatment alone. These results can be seen in Figure 6. Of note is the overall slope of the interlaced treatment group plot, which is steeper than that of either single- agent treatment group, especially from 24 h to 48 h. These results indicate that interlacing the chemothera- peutic drug paclitaxel with T-type calcium channel blocker NNC-55-0396 is effective at impeding the growth of MCF-7 breast cancer cells in a manner that may be synergistic. 3.6. Interlaced: In Vivo The success of the interlaced treatment scheme in vitro justified a similar approach in an in vivo model. Athy- micovarectemized female nude mice were implanted Copyright © 2013 SciRes. JCT  Exploiting MCF-7 Cells’ Calcium Dependence with Interlaced Therapy 38 Figure 6. WST-8 assay of MCF-7 cells treated with NNC- 55-0396, paclitaxel, or both in alternation. MCF-7 cells treated in vitro with an alternating schedule of NNC-55- 0396, paclitaxel, and NNC-55-0396 (24 h each) were signifi- cantly inhibited in growth, compared to cells treated with either NNC-55-0396 alone or paclitaxel alone for 72 h. Data normalized to that of untreated control cells at the same time point. ANOVA with Bonferonni multiple comparison post-tests, #p < 0.05 vs. NNC-55-0396, *p < 0.05 vs. pacli- taxel. with subcutaneous slow-release estradiol pellets, fol- lowed by being implanted with MCF-7 cell xenografts in the mammary fat pads. After tumors were established, animals were randomly segregated into groups and treat- ed with mibefradil, paclitaxel, or interlaced mibefradil/ paclitaxel in a weekly treatment regimen. Mibefradil was administered orally for 3 consecutive days in a given week; paclitaxel was administered via intraperitoneal injection once in a given week. Single-agent treatment groups received one of these; the interlaced group re- ceived both. The tumors started out very similar in size. Over the course of the 35 days of the experiment, the mice given interlaced therapy were the only animals whose tumors became significantly smaller than those in mice that received no treatment. The plot of tumor size vs. time can be seen in Figure 7. While the control and single-agent treatment groups’ tumors are steadily grow- ing, the interlaced treatment groups’ tumors are held in check. For days 30, 32 and 35 the interlaced group’s tu- mors are significantly smaller than those of the control group. Figure 8 shows the close correlation of column plots of final tumor volume (Day 35) and tumor weight as determined immediately following animal sacrifice and tumor excision. 4. Discussion This study demonstrates the importance of T-type cal- cium channels in breast cancer cell calcium signaling and growth. Both extracellular calcium and intracellular cal- cium stores are crucial for MCF-7 cell growth. T-type calcium channels play a significant role in the manage- ment of MCF-7 intracellular calcium concentration. In- hibition of T-type calcium channels prevents these breast cancer cells from passing through the phases of the cell Figure 7. In vivo MCF-7 xenograft growth inhibition by interlaced mibefradil and paclitaxel. MCF-7 tumor xeno- grafts in nude mice experienced significant growth inhibi- tion from a treatment schedule that alternated mibefradil and paclitaxel when compared to control or either mibe- fradil alone or paclitaxel alone. Figure 8. Final tumor volume and weight comparison. MCF-7 tumors were removed from mice after completion of the in vivo interlaced treatment study and weighed. The tumor weight data closely resembles the final tumor volume measurement data. cycle. This dependence makes T-type calcium channels a potential target in breast cancer therapy, through the ef- fects of combining T-type calcium channel blockers with chemotherapy. The importance of calcium to MCF-7 cell proliferation is demonstrated here. MCF-7 breast cancer cells maintain a basal average intracellular calcium concentration high- er than that maintained in non-tumorigenic MCF-10A cells, perhaps allowing for enhanced calcium signaling. Reduction in calcium available for signaling, through either chelating extracellular calcium or depleting intra- cellular stores, significantly reduces the growth of MCF- Copyright © 2013 SciRes. JCT  Exploiting MCF-7 Cells’ Calcium Dependence with Interlaced Therapy 39 7 cells. A cell expressing T-type calcium channels has an extra pathway for calcium ion entry into the cytosol from the extracellular fluid, and so may be expected to have an elevated intracellular calcium concentration at rest due to the properties of these channels. T-type calcium channels have a low threshold for activation, which would allow for channel conductance at transmembrane voltages near normal resting potential [16]. Blocking T-type calcium channels with mibefradil reduces MCF-7 intracellular calcium concentration, confirming the role that these channels play in calcium concentration maintenance. Cancer cells have been reported to be relatively insensi- tive to reductions in extracellular calcium concentration [17]. The results reported here indicate that extracellular calcium concentration can affect MCF-7 cell prolifera- tion, but only when reduced by nearly two orders of magnitude. Calcium signaling may be altered in MCF-7 cells as demonstrated by elevated basal concentration and aberrant channel expression, but it certainly is still nec- essary. Depletion of endoplasmic reticulum calcium stores by thapsigargin is known to exacerbate the endo- plasmic reticulum stress response in MCF-7 cells [18], and prolonged exposure to thapsigargin is known to in- duce chromosomal fragmentation in MCF-7 cells [19]. Activating the endoplasmic reticulum stress response via thapsigargin mediated endoplasmic reticulum calcium store depletion may help explain the time- and dose-de- pendent reduction in MCF-7 cell growth seen in this study. Perhaps T-type calcium channels serve as an aux- iliary conduit in MCF-7 cells for calcium ions to be moved from the extracellular fluid through the cytosol and into intracellular stores, to serve as insurance against induction of endoplasmic reticulum stress responses. The inhibitory effects of T-type calcium channel bloc- kade on the proliferation of MCF-7 cells has been dem- onstrated previously [7]. The reduction in intracellular calcium concentration, an immediate consequence of T- type calcium channel blockers, is demonstrated here. Mibefradil significantly reduces MCF-7 cell average intracellular calcium concentration. However, exactly how that change in free calcium ions results in a growth reduction is unclear. Mibefradil is known to upregulate p21 expression in esophageal cancer cells, a finding con- firmed with RNA interference [8]. Presumably, a similar “end result” of growth regulating signaling initiated by reduced T-type calcium channel activity may be detect- able in MCF-7 cells, but there remain many unknown steps between T-type calcium current and cell cycle regulation. Further investigation of the mechanisms in- volved in these phenomena is needed. Application of NNC-55-0396 in conjunction with pa- clitaxel in an interlaced treatment scheme is more effec- tive at reducing MCF-7 cell number than either NNC- 55-0396 or paclitaxel alone. These results can be viewed in light of our other findings. By halting cells at the G1/S checkpoint, the first 24 h of NNC-55-0396 application lowers MCF-7 cell number relative to control and re- duces the population’s cell cycle heterogeneity. This re- duction in heterogeneity makes the cells more susceptible to paclitaxel, which kills them as they pass through the remainder of the cell cycle. This illustrates the potential of T-type calcium channels as a target in cancer therapy. Our in vitro results led us to apply interlaced therapy in an in vivo model. MCF-7 tumor xenografts were es- tablished in nude athymic female mice and tumor size was monitored via caliper measurements. While all tu- mors initially are of very similar size, separation is seen at the termination of the experiment. In the last week, the tumors of the mice receiving interlaced therapy are sig- nificantly smaller than tumors in control mice to a statis- tically significant degree; the sizes of tumors in animals receiving either mibefradil alone or paclitaxel alone were not found to be significantly different from those of con- trol animal tumors. That the interlaced treatment suc- ceeded in vivo to a degree larger than traditional chemo- therapy alone is in accord with our in vitro results and our hypothesis. Mibefradil is an effective T-type calcium channel blocker but it also blocks L-type calcium chan- nels via an intracellular metabolite [20]. While this is not a problem in vitro, as MCF-7 cells do not express L-type calcium channels, this is a matter of concern in terms of organismal physiology. L-type calcium channels are cri- tical in the function of tissues such as cardiac muscle and vascular smooth muscle. We did not observe any signifi- cant effects on animal body weight, pulse or blood pres- sure as a result of mibefradil administration (data not shown), but side effects are a relevant concern. Further investigation of highly specific channel blockers such as NNC-55-0396 is important to realizing the potential of targeting T-type calcium channels in clinical cancer the- rapy. REFERENCES [1] H. L. Roderick and S. J. Cook, “Ca2+ Signaling Check- points in Cancer: Remodeling Ca2+ for Cancer Cell Pro- liferation and Survivial,” Nature Reviews Cancer, Vol. 8, No. 5, 2008, pp. 361-375. doi:10.1038/nrc2374 [2] D. Walker, T. Sun, S. MacNeil and R. Smallwood, “Mod- eling the Effect of Exogenous Calcium on Keratinocyte and HaCat Cell Proliferation and Differentiation Using and Agent-Based Computational Paradigm,” Tissue En- gineering, Vol. 12, No. 8, 2006, pp. 2301-2309. doi:10.1089/ten.2006.12.2301 [3] S. Rosenberger, I. S. Thorey, S. Werner and P. Boukamp, “A Novel Regulator of Telomerase. S100A8 Mediates Differentiation-Dependent and Calcium-Induced Inhibi- tion of Telomerase Activity in the Human Epidermal Keratinocyte Line HaCaT,” The Journal of Biological Copyright © 2013 SciRes. JCT  Exploiting MCF-7 Cells’ Calcium Dependence with Interlaced Therapy Copyright © 2013 SciRes. JCT 40 Chemistry, Vol. 282, No. 9, 2007, pp. 6126-6135. doi:10.1074/jbc.M610529200 [4] G. Legrand, S. Humez, C. Slomianny, E. Dewailly, F. Vanden Abeele, P. Mariot, F. Wuytack and N. Prevar- skaya, “Ca2+ Pools and Cell Growth. Evidence for Sar- coendoplasmic Ca2+-ATPases 2B Involvement in Human Prostate Cancer Cell Growth Control,” The Journal of Biological Chemistry, Vol. 276, No. 50, 2001, pp. 47609- 47614. doi:10.1074/jbc.M107011200 [5] V. Lehen’kyi, M. Flourakis, R. Skryma and N. Prevar- skaya, “TRPV6 Channel Controls Prostate Cancer Cell Proliferation via Ca(2+)/NFAT-Dependent Pathways,” Oncogene, Vol. 26, No. 52, 2007, pp. 7380-7385. doi:10.1038/sj.onc.1210545 [6] J. T. Taylor, X. B. Zeng, J. E. Pottle, K. Lee, A. R. Wang, S. G. Yi, J. A. S. Scruggs, S. S. Sikka and M. Li, “Cal- cium Signaling and T-Type Calcium Channels in Cancer Cell Cycling,” World Journal of Gastroenterology, Vol. 14, No. 32, 2008, pp. 4984-4991. doi:10.3748/wjg.14.4984 [7] J. T. Taylor, L. Huang, J. E. Pottle, K. Liu, Y. Yang, X. Zeng, B. M. Keyser, K. C. Agrawal, J. B. Hansen and M. Li, “Selective Blockade of T-Type Ca2+ Channels Sup- presses Human Breast Cancer Cell Proliferation,” Cancer Letters, Vol. 267, No. 1, 2008, pp. 116-124. doi:10.1016/j.canlet.2008.03.032 [8] F. Lu, H. Chen, C. Zhou, S. Liu, M. Guo, P. Chen, H. Zhuang, D. Xie and S. Wu, “T-Type Ca(2+) Channel Ex- pression in Human Esophageal Carcinomas: A Functional Role in Proliferation,” Cell Calcium, Vol. 43, No. 1, 2008, pp. 49-58. doi:10.1016/j.ceca.2007.03.006 [9] G. E. Bertolesi, C. Shi, L. Elbaum, C. Jollimore, G. Ro- zenberg, S. Barnes and M. E. M. Kelly, “The Ca2+ Chan- nel Antagonists Mibefradil and Pimozide Inhibit Cell Growth via Different Cytotoxic Mechanisms,” Molecular Pharmacology, Vol. 62, No. 2, 2002, pp. 210-219. doi:10.1124/mol.62.2.210 [10] A. Panner and R. D. Wurster, “T-Type Calcium Channels and Tumor Proliferation,” Cell Calcium, Vol. 40, No. 2, 2006, pp. 253-259. doi:10.1016/j.ceca.2006.04.029 [11] W. Li, S. L. Zhang, N. Wang, B. B. Zhang and M. Li, “Blockade of T-Type Ca2+ Channels Inhibits Human Ovarian Cancer Cell Proliferation,” Cancer Investigation, Vol. 29, No. 5, 2011, pp. 339-246. doi:10.3109/07357907.2011.568565 [12] A. Panner, L. L. Cribbs, G. M. Zainelli, T. C. Origitano, S. Singh and R. D. Wurster, “Variation of T-Type Calcium Channel Protein Expression Affects Cell Division of Cul- tured Tumor Cells,” Cell Calcium, Vol. 37, No. 2, 2005, pp. 105-119. doi:10.1016/j.ceca.2004.07.002 [13] P. Mariot, K. Vanoverberghe, N. Lalevee, M. F. Rossier and N. Prevarskaya, “Overexpression of an Alpha 1H (Cav3.2) T-Type Calcium Channel during Neuroendo- crine Differentiation of Human Prostate Cancer Cells,” Journal of Biological Chemistry, Vol. 277, 2002, pp. 10824-10833. doi:10.1074/jbc.M108754200 [14] Y. Q. Wang, G. Brooks, C. B. Zhu, W. Z. Yuan, Y. Q. Li, and X. S. Wu, “Functional Analysis of the Human T- Type Calcium Channel Alpha 1H Subunit Gene in Cel- lular Proliferation,” Journal of Genetics & Genomics, Vol. 29, No. 8, 2002, pp. 659-665. [15] J. F. Whitfield, A. L. Boynton, J. P. MacManus, R. H. Rixon, M. Sikorska, B. Tsang, P. R. Walker and S. H. Swierenga, “The Roles of Calcium and Cyclic AMP in Cell Proliferation,” Annals of the New York Academy of Sciences, Vol. 339, No. 1, 1980, pp. 216-240. doi:10.1111/j.1749-6632.1980.tb15980.x [16] V. Crunelli, T. I. Toth, D. W. Cope, K. L. Blethyn and S. W. Hughes, “The ‘Window’ T-Type Current in Brain Dy- namics of Different Behavioural States,” Journal of Phy- siology, Vol. 562, 2005, pp. 121-129. doi:10.1113/jphysiol.2004.076273 [17] J. F. Whitfield, “Calcium Signals and Cancer,” Critical Reviews in Oncogenesis, Vol. 3, No. 1-2, 1992, pp. 55-90. [18] N. Dejeans, N. Tajeddine, R. Beck, J. Verrax, H. Taper, P. Gailly and P. B. Calderon, “Endoplasmic Reticulum Cal- cium Release Potentiates the ER Stress and Cell Death Caused by an Oxidative Stress in MCF-7 Cells,” Bio- chemical Pharmacology, Vol. 79, No. 9, 2010, pp. 1221- 1230. doi:10.1016/j.bcp.2009.12.009 [19] C. Jackisch, H. A. Hahm, B. Tombal, D. McCloskey, K. Butash, N. E. Davidson and S. R. Denmeande, “Delayed Micromolar Elevation in Intracellular Calcium Precedes Induction of Apoptosis in Thapsigargin-treated Breast Cancer Cells,” Clinical Cancer Research, Vol. 6, 2000, pp. 2844-2850. [20] S. Wu, M. Zhang, P. A. Vest, A. Bhattacharjee, L. Liu and M. Li, “A Mibefradil Metabolite Is a Potent Intracel- lular Blocker of L-Type Ca2+ Currents in Pancreatic β-Cells,” The Journal of Pharmacology and Experimental Therapeutics, Vol. 292, No. 3, 2000, pp. 939-943.
|