An experimental characterization of the Van der Waals forces involved in volatile organic compounds (VOC) dissolved into stationary phases of gas liquid chromatography (GLC) has been started at the beginning of the seventies. This field has been reactivated from 1994 thanks to a fruitful cooperation between our CNRS team and the group of Ervin Kováts at the
Federal Polytechnic School of Lausanne. The applied strategy can be summarized, in the first instance, as the experimental measurement of accurate and superabundant mutual affinities of a limited number of VOC and stationary phases and their processing using an original tool named Multiplicative Matrix Analysis (MMA). Then, in the second stage, the obtained results have been compared with molecular properties well established, as the Van der Waals molecular volume, the refraction index and the polar surface area (PSA), in order to get generalized values for any compound. The present study summarizes the positive results developed in our three last papers on this topic (2013, 2016 and 2018), as well as the attempt to overcome the negative ones using enthalpies of vaporization.
Enthalpy of Vaporization Van der Waals Intermolecular Forces Molecular Polarity Polar Surface Area Gas-Liquid Chromatography Chemo-Informatics Olfaction1. Introduction
One of our activity axes for various decades in our CNRS group and personally recently pursued, has been to characterize the intermolecular forces involved in solute-solvent interactions. This, in the purpose of getting accurate numerical values reflects these forces. We have preferred until now the GLC (Gas-Liquid Chromatography) approach, but the purpose of the present study is to explore, possibly more fruitfully, the enthalpy of vaporization splitting.
1.1. Principal Positive Results and Some Drawbacks of the GLC Approach
It has been recently published two papers on the intermolecular forces involved in GLC, respectively focused on solutes and on stationary phases acting as solvents [1] [2]. They correspond to the state of art of a thematic field started in 1972 [3] involving the derivation of solute solvation parameters or descriptors from retention indices on quite a few selected chromatographic columns, in order to attempt QSAR (Quantitative Structure Activity Relationships) with olfactory properties. These two recent publications, however, are strictly limited to the physicochemical aspects of the topic. From 1976, as suggested by Eon [4] and applied by Karger et al. [5] [6], our group has taken into account five solute solvation parameters instead of four as previously [7]. Three of these five parameters are related to the Van der Waals forces and the two others to the hydrogen bonding, as summarized in Figure 1.
1.1.1. Strategy Adopted since 1972
It can be summarized in three steps:
· Establishment of accurate and superabundant databases of retention indices in GLC. Let us highlight the importance of expressing the retention in terms of Kováts indices (RI), the only one mapping out the observation of Rohrschneider [8] in Equation (1), i.e. that can be expressed as a matricial product of solvation parameters of solutes and solvents:
RI − RI CH4 = δ D + ω W + ε E + α A + β B (1)
where the acronyms are those of Figure 1, RICH4 being the retention index of methane, always equal to 100.
The first database we used in 1972 and 1976 [3] [7] is an unpublished matrix of retention indices of 75 solutes on 25 stationary phases provided by McReynolds
[9], in a perspective of a further cooperation. McReynolds itself recognized that in this database the property of proton donor was poorly represented among the 25 phases and started to complete it with a 26th phase, the Hyprose SP-80. We did not hear about the achievement of this plan (McReynolds deceased in 1976).
Shortly after our publication [7], a fruitful cooperation has gradually been established between our CNRS team and the group of Ervin Kováts at the Federal Polytechnic School of Lausanne (EPFL). This cooperation has stepped up in 1994, when the Kováts group provided us a new and accurate matrix R including a suitable proton donor phase and two paraffin phases of different molecular weight phases, these two latter permitting an accurate determination of the ε solute parameter (133 solutes × 10 phases in the first version [10]). After a new study on an 11th phase [11] and a controversy on the apolar phases [12], a revised and refined version of this matrix R was adopted (127 solutes × 11 phases) [13]. Let us specify that all these phases have been specially synthesized by the Kováts group and that they are not commercially available (e.g. [10] for the two paraffin phases and [14] for the 8 first polar phases).
· Multiplicative Matrix Analysis (MMA). The operating principle of this data processing, initially developed in cooperation with Robin [15], is specified in [10] and the updated version of its transcript in MatLab language is available in [13]. As it can be seen in Figure 2, this analysis can be considered as a tool for testing independently the validities of each one of the elements of the input matrix A (here, the solute descriptors according to various semi-theoretical approaches), applied to an experimental matrix R, supposed to be the product of matrices A and B (here the experimental matrices of retention indices). In addition, the MMA algorithm can provide an improvement of the matrix A (consequence of the superabundant information), and propose a matrix B (here related to the solvents). In contrast, the reproducibility of the matrix R is totally independent of the validity of the input matrix A elements, and only depends of their number (here fixed to 5).
· Attempts of extending the accurate obtained results to a larger number of compounds. Two avenues have been explored. The first one is the experimental method using five selected stationary phases in filled columns. It could be quite easy to consider an automated device including five columns in parallel. In the publication [13] it is described a method for selecting a suitable set of phases from those commercially available, in order to avoid disappointing results as ours in 1982 for 240 solutes [16].
The second avenue we have explored for extending the accurate results get for a few number of compounds is a Simplified Molecular Topology (SMT). This tool will be summarized in the Materials and Methods section since it is again applied in the present study, in spite of various relatively unsatisfactory results in the past [17] [18].
1.1.2. Achieved Results
They are detailed in [1] for solutes (VOC, i.e. volatile organic compounds), and in [2] for solvents (i.e. gas liquid chromatographic stationary phases). Without entering in too much details, they can be summarized as follows:
· The induction-polarizability descriptor ε of solutes. This descriptor, or parameter named ε as reminiscent of electron, has firstly be drawn in previous studies [19] [20] [21] [22] on the basis of a high value of the refractive index for a given molar volume. From 1976 until 2016, these versions have been improved using the MMA processing applied to the superabundant GLC experimental data of McReynolds [9] and of Kováts and co-workers [10] [11] [12] [23] [24] [25] [26]. The definition of ε in its most recently published version according to [1] is as follows:
ε 2016 = 10 f n R V W − 2.828 V W + 62.67 (2)
with: f n R = ( n R 2 − 1 n R 2 + 2 ) (3)
in which nR stand for the refractive index at 20˚C, and Vw for the intrinsic molecular volume according to Molinspiration [27].
Because the refractive index is not always available in liquid state, an alternative expression of ε2016 has been proposed on the basis of 14 molecular features which can be named ε2016-SMT (see its complete definition on the bottom of Figure 3 in [1]). The correlation coefficient between ε2016 and ε2016-SMT has been found equal to 0.978 for 447 VOC in liquid state at room temperature.
· The dispersion descriptor δ of solutes. Similarly, in its more recent expression the δ2016 descriptor has been taken from the product fnRVw, and in its alternative version δ2016-SMT from an equation including 10 molecular features (see its complete definition on the top of Figure 3 in [1]). The correlation coefficient between δ2016 and δ2016-SMT has been found equal to 0.997 for 447 VOC in liquid state at room temperature.
· The solute descriptors α and β involved in hydrogen bonding. The solute parameters of acidity and basicity according to Abraham in 1993 [28], as elements of an input matrix A submitted to the MMA processing on the same matrices R of retention indices as for ε, remain almost identical to the output values and therefore validated. In contrast, the extension of these parameters to a greater amount of compounds using the SMT procedure has failed each time an intramolecular hydrogen bonding was suspected. The solution could be a molecular topology extended to the first neighbors of each atom of a given molecule, which would require many more learning data then the available ones. Our personal conclusion is a limitation, perhaps temporary, of our attempt to extend the three parameters related to the Van der Waals forces.
· The orientation descriptor ω of solutes. As shown in Figure 3, we have obtained for the first time in 2016 [1] a satisfactory expression of the orientation descriptor ω (named ω2016), as a function of 11 molecular features.
At this stage, it should however be underlined that this expression of ω, based on the results shown in Figure 3, presents a lack of connection to other well established molecular properties, on the contrary to δ and ε. One of the goals of the present study is to try to make progress on it.
· The solvent parameters. The only accurate and available database of solvent parameters involved in Van der Waals forces we have found appears noticeably smaller than for solutes. It is limited to W and E for 11 stationary phases, as it can be seen in Table 1. Indeed, the parameter D is a constant as a direct consequence of the retention indices definition, which is a relative expression to n-alkanes of the solute-solvent affinities, and not an absolute expression. That implies the taking into account of an additional solvent parameter, named b by McReynolds [29] and defined as the linear regression slope of the logarithm of the net retention times of n-alkanes, versus their number of carbon atoms. In order to avoid any confusion, we propose to name as for now McR-b this parameter.
We can see on the present Table 1 that not only D is almost a constant, but also McR-b. This latter phenomenon is due to the nature of molecular structures selected by the Kováts group. Fortunately, McReynolds published McR-b experimental values for 226 filled columns (207 phases with range values of 0.16 - 0.29) [29]. We have been able to chemically identify 68 of these phases, the remaining ones being commercial reproducible polymers or mixtures but not chemically defined compounds.
The three main observations to be retained about the solvent parameters as they are summarized in [2] are:
- Their prominent proximity with the Polar Surface Area (PSA), particularly for McR-b, but not only. Therefore PSA, promoted as of pharmacological interest ( [30] [31] [32] [33] among many other ones), also deserves now a physicochemical consideration.
- All the molecular features, including PSA, involved in a prediction model of solvent solvation parameters of GLC stationary phases should be divided by their intrinsic molecular volume on the contrary to that observed for solutes [2] [34].
- The observation by the Kováts group [35] [36] [37] that the n-alkane stationary phases of low molecular weight display a polar behavior of induction is totally confirmed [2] [34].
Solvent descriptors D, W and E of the Van der Waals forces involved in GLC according to [<xref ref-type="bibr" rid="scirp.99955-ref13">13</xref>], and McReynolds b parameter according to [<xref ref-type="bibr" rid="scirp.99955-ref10">10</xref>] for 11 stationary phases studied by the Kováts group (Synthetic table 1 from [<xref ref-type="bibr" rid="scirp.99955-ref2">2</xref>]). See text
Let us mention an erratum in the Table 2 of [10] concerning the two first lines: partial inversion of data between the two apolar phases, for which only the McR-b values are right. In contrast all the values in Table 4 of [13], in Table 1 of [2] and in Table 1 of the present study have been verified and are correct.
1.2. Principal Question Arising on the Mutual Consistency of Our Three Last Publications
As mentioned above, the main observed difference between solute and solvent parameters is that the former can be expressed as functions of one or various molecular features, whereas the latter as functions of similar features but divided by their intrinsic molecular volume. This finding may be seen slightly surprising but a priori acceptable.
The main question comes from our publication of 2013 [38] on a different context: i.e. a quantitative structure activity relationship (QSAR) in olfaction. We have retained an olfactory property that we have named comfort OLPI (OLfactory Perceived Intensity). Anyone has observed that in vision there is an optimal luminance, not too high not too low, for a comfortable lecture. There is a similar phenomenon in the Honey-Bee for its learning of association between the sweet tasting and a given olfactory perception. It has been shown in 1989 and 1990 that the optimal learning is observed when an elicited electroantennogram (EAG) reflecting the OLPI is equal to 1 - 1.2 mV, everything being equal in the experimentation procedure. For example, olfactory stimulations eliciting EAG equal to 0.5 or 2.5 mV result in an appreciable deterioration of the learning, whatever their nature are [39] [40] [41] [42].
On the other hand, it has also be shown in 1989 by Patte et al. [43] that stimulus-EAG response curves in the Honey Bee display a convergence point, as shown in Figure 4, for three of the odorants studied by these authors, as examples.
Because the superimposition of the 59 odorant curves on the same diagram would be difficult to interpret, these authors have parameterized the experimental points for each of the 59 odorants, using the Hill model [45] reported in Equation (4).
R = ( C / C x ) n R M 1 + ( C / C x ) n (4)
in which R stands for the electroantennogram amplitude of response, RM for the maximal amplitude, C for the concentration, n for the power law exponent and Cx for the concentration corresponding to the inflexion point in the log-linear sigmoid curve.
This model appears often suitable in life sciences, each time the experimental curves present a sigmoid shape in linear-log coordinates and a hyperbolic shape in log-log coordinates, as it is the case in Figure 4.
It can also be considered an anchor point (C0, R0) in the bottom of the slanted almost straight line of the log-log drawing as in Figure 4, given by the Equation (5) derived from (4):
log C 0 = 1 n log R 0 R M − R 0 + log C X (5)
One of the Hill model main advantages is that it easily allows iterative procedures resulting to an optimal fitting between experimental points and curvilinear drawings like in Figure 4, only based on the four biological parameters mentioned above: n, RM, CX and C0. (In the Patte et al. experimentation on the Honey Bee, R0 has been fixed to 0.1 mV, as the minimal electrophysiological value distinct from the base-line).
That has allowed Patte et al. to characterize the 59 studied odorants using these four olfactory parameters and to observe two things: 1) their strong mutual correlations and 2) high improvements of these correlations when the concentrations are expressed in fractions of saturated vapor pressure, as reflected by Equation (6) and Equation (7):
log C x = log SVP + log R M n − 2.33 ( r = 0.99 ; N = 59 ; F = 946 ; without outliers) (6)
log C 0 = log SVP + log R 0 n − 2.33 ( r = 0.97 ; N = 59 ; F = 404 ; without outliers) (7)
in which SVP stands for the saturated vapor pressure expressed in bars and the constant −2.33 as the abscissa CC of the convergent (and comfort) point.
It should be underlined that this type of statistical results is unusual in life sciences.
In fact, the original Equation (7) from [43] should theoretically be completed as follows:
log C 0 = log SVP + log R 0 n − log R C n − 2.33 (8)
in which RC stands for the response at the convergence point, and that by chance in the present case, as it can be verified in Figure 4, RC = 1 mV, and therefore logRC/n = 0
Without deeply entering into the physical chemistry of solutions, the role played by the saturated vapor pressure at very different concentrations consists to say that the odorants reach the olfactory dendrites through a dry route into pores-tubules until the immediate proximity of the olfactory cilia, as it is shown in various anatomic studies on Insects [46]. Of course, because the depolarization of membrane cells implies an ionic exchange, a minimum of water has to be present at the surface of dendrites cilia. The water layer could, however, be thin enough to make “ideal” (in the sense of Raoult law) the behavior of the whole plasma membrane.
Furthermore, the high mutual correlations between the olfactory parameters CX, RM and n, or C0, R0 and n, respectively observed in the Equation (6) and Equation (7), reflect the intersection of the 59 curves for the abscissa −2.33 of the log fraction of saturated vapour pressure, as it is exemplified in Figure 4 (i.e. for a fraction of SVP near to 0.005).
From psychophysical studies (i.e. human responses), it has also be shown for a long time [21] [47] significant mutual correlations between the olfactory thresholds C0 and the power law exponents n. This has been partially interpreted as the fact that the olfactory dendrites of neuroreceptors in Vertebrates are immersed in olfactory aqueous mucus at least 30 times thicker than the water layer in Insects mentioned above, where the VOC (volatile organic compounds) can not obey to the Raoult law. We have proposed in 2013 [38] a polar expression so-called vertolf (as Vertebrates Olfactory Filter) supposed to play in the human olfactory mucus and perhaps in all Vertebrates, an analogous role as the saturated vapor pressure in Insects olfaction, and also more effective than those previously applied [21] [47]. The proposed definition of vertolf is as follows:
v e r t o l f = 7.406 MR 100 + 3.604 PSA2 V w (9)
in which MR stands for the molar refraction, Vw for the Van der Waals molar volume and PSA2 for one of the slightly modified expressions of the original polar surface area, specified in the Material and Methods section.
The equation analogous to (7) valid for psychophysical data, as it appears in 2013 [38], is:
log C 0 = − v e r t o l f − 0.8177 n − 2.3 ( r = 0.77 , N = 186 , F = 89 , without outliers) (10)
At this stage, in spite of the moderate value of the resulting correlation coefficient with the Equation (10), the analogy between Equation (7) and Equation (10) must be underlined. It however remains a difficulty concerning the definition of vertolf in Equation (9), which appears as the sum of two terms: one as suitable for solutes (7.406MR/100) and another one as suitable for solvents (3.604PSA2/Vw). Indeed, as seen in §1.1.2, molecular features (here MR and PSA2) have to be divided by their intrinsic molecular volume only for solvents. The principal purpose of this study is to overcome this apparent inconsistency in definition of vertolf.
2. Materials and Methods2.1. Statistical Tools
In addition to the Microsoft Excel Windows facilities for drawing diagrams and handling data sets, the SYSTAT 12® for Windows has been applied for stepwise MLRA (Multidimensional Linear Regression Analysis).
2.2. Simplified Molecular Topology (SMT)
The principle of this tool has already be presented and detailed elsewhere [17] [18]. The version used here is similar to that in [1]. It only takes into account, for each atom of a molecule, its nature and the nature of its bonds, leaving aside the nature of its first neighbors, with the exception of amines linked to a carbon linked itself to O2. This exception occurs in amides. Each atom is provided with an index comprising a series of digits. Their sum is at most equal to its valence. The value of the digits define the type of bonds (1 for a single, 2 for a double bond, etc.), but the bonds with hydrogen are excluded. So, the only possibilities for oxygen, for example, are O1 (alcohols, carboxylic acids), O11 (ethers, esters, lactones) and O2 (ketones, aldehydes, esters, carboxylic acids, nitro compounds, lactones). Only are considered in the present study the atoms C, H, O, N, P, S, F, Cl, Br, I. However, the compounds including a given atom only linked to hydrogen (e.g. CH4, OH2, NH3, SH2) are excluded. In addition, a connectivity parameter due to Zamora [48] called the “smallest set of smallest rings” (SSSR) is taken into account. According to this concept, for the naphthalene for example which contain two individual C-6 rings and one C-10 ring embracing them, only the two six numbered rings are considered. Two six numbered rings corresponding to 12 carbon atoms, the SSSR value of naphthalene is therefore be taken equal to 12.
Let us specify that the calculations using the SMT procedure have been made manually in this study, using 2D molecular drawings from ChemSpider [49].
2.3. Global Spherical Surface
One of the ways to overcome the apparent contradiction between our three last publications mentioned in the Introduction could be at the first sight the use of molecular surface area instead of the molecular volume. In the presentation of the Molecular Surface Area Plugin by Chemaxon, it is specified that two types of available molecular surface area calculations are available: the Van der Waals surface area and its Solvent Accessible Surface Area (SASA), both being expressed in Ǻ2 [50]. The calculations are based on Ferrara et al. [51]. We did not have yet the opportunity to apply and test the sophisticated SASA tool to our GLC (Gas Liquid Chromatographic) experimental data, but it seems that its applicability has been thought for different solute/solvent situations different of ours, for example proteins as solutes and water as solvent, on the contrary to that applied in GLC (i.e. volatile organic compounds as solutes and stationary phases as solvents). By contrast, we tested the Van der Waals molecular surface instead of Vw (both from Chemaxon [50]), in our olfactory QSAR study of 2013 [38]) and found very similar results.
Nevertheless, we have tested in the present study the property we propose to call the Global Spherical Surface (GSS), expressed as derived from the molecular volume as follows:
GSS = ( 36 π ) 1 / 3 V w 2 / 3 ≡ 4.836 V w 2 / 3 (in Ǻ2 when Vw is expressed in cubic angstroms) (11)
This expression means that solute molecules are considered as small spheres disseminated in the solvent.
2.4. Sources of Data Values for Well Established Molecular Properties2.4.1. Polar Surface Area
We have considered until now three variants of PSA:
- The most classical, only including the polar atoms N and O. We have selected the values named TPSA (T as topological) established by Molinspiration [27], and in turn renamed here PSA1.
- The variant including the same polar atoms N and O as in TPSA, but also the various divalent S according to Ertl et al. [33]. This expression has been adopted by ChemSpider [49], without decimal. We name it PSA3.
- In 2013 we have named PSA2 a third variant initially identical to PSA3, but diminished of the pentavalent N present in nitrates according to [33], since this molecular feature cannot be considered as polar (more details in [38]). PSA2, as specified in the Equation (9), has been selected by the MLRA processing as the most suitable variant in a QSAR olfactory application.
All the sources of PSA come from Molinspiration [27], modulated by [33].
2.4.2. Intrinsic Molecular Volume
We have adopted in the present study the values of molecular volume according to Molinspiration [27]. We name it Vw (w as Van der Waals), in order to avoid any confusion with V20, the molecular volume at 20˚C (molar mass/density) which is not an additive property [52] [53].
2.4.3. Enthalpy of Vaporization, Refractive Index, Molar Refraction, Boiling Point, Molecular Volume at 20˚C and Possible Other Properties
Most of those applied here are from ChemSpider [49] and from the Handbook of Chemistry and Physics [54].
3. Results3.1. Shortened Characterization Completely or Partially Achieved at the Beginning of the Present Study, for the Descriptors Reflecting the Van der Waals Forces Involved in Solutions
1) The two descriptors of solutes δ and ε (see meaning of acronyms in Figure 1). Both are satisfactorily characterized via a combination of well established molecular properties, i.e. refractive index at 20˚C (nR) and Van der Waals molecular volume (Vw), according to the Equation (12) and Equation (13) from [1]:
δ 2016 = f n R V w with f n = n R 2 − 1 n R 2 + 2 (12)
ε 2016 = 10 f n R V w − 2.828 V w + 62.67 (13)
In addition to these definitions, named “theoretical” in [1], these two descriptors have also been defined in a similar way to the descriptor ω in Figure 3, i.e. using around a dozen of molecular features permitting an acceptable prediction of the descriptors experimentally obtained and published in [13].
2) The descriptors of solvents. The predicted W and E descriptors in [2] using a SMT procedure have spectacularly well matched those experimentally established in [13] (r = 0.9997). However it must be underlined that these results have been obtained using only 11 stationary phases. Their principal interest is the confirmation of the polar behavior of the supposed apolar phases of low molecular weight (E), and also that the strength of the inductive power depends, wholly or in part, upon the strength of the hydroxyl function (also E). In addition, the importance of the fluorine in the strong polarity phases is underlined (W).
Otherwise, two updated predictions of the McR-b descriptor using PSA (unpublished until now) are given for the database on the study (75 phases on 86 columns), by the Equation (14) and Equation (15)):
McR-b 2019 = 0.299 − 0.253 PSA V W r = 0.946 , F = 710 (14)
McR-b 2019 = 0.302 − 0.229 PSA V W − 0.992 O11 V W r = 0.959 , F = 472 (15)
[ F = 548 ] [ F = 26 ]
whatever the chosen molecular features are (e.g. with or without PSA, with 66 columns as in 2011 or with 86 as presently), all the higher tests values of validation are obtained with the ratio molecular feature over Vw. And when it is included with other features, PSA always plays a central role in the regressions.
Let us note that variants of PSA mentioned in the Material and Methods section are equally validated in the Equation (14) and Equation (15), since among the phases selected in the database of McReynolds no one includes sulfur nor nitro compounds.
3) The ω descriptor for solutes. As it results from the Introduction, the principal effort remaining to do in the present topic is related to an improvement in the characterization of the ω descriptor for solutes.
3.2. Comparative Performances of Various Predictability Models of the ω Descriptor
As a last development of our GLC approach, in Figure 5 are summarized some comparisons of performances of four models including the one already published in 2016 [1] and reproduced in Figure 3. The four of them can be considered as rather good, but the one called ω2020-SMT seems clearly preferable to the one including Vw. Indeed, in addition of the absence of outlier, the ω2020-SMT model appears preferable to the ω2016-SMT one on the basis of a visual comparison of the two correspondent correlograms in Figure 3 and Figure 6: the experimental points in Figure 6 seem to be more regularly distributed.
3.3. The Enthalpy of Vaporization Approach for Compounds in Liquid State at Room Temperature
The results obtained using the enthalpy of vaporization have been based on a databank of 445 volatile organic compounds (VOC) in liquid state at room temperature (more often at 20˚C), chosen as including numerous chemical functions: alcohols, aldehydes, ketones, carboxylic acids, ethers, amides, lactones, nitro-compounds, nitriles, amines, esters, various types of halogen and sulfur compounds and hydrocarbons, both of saturated or unsaturated types, with and without mono and polycycles. A similar set of 447 VOC was used in 2016 [1], from which have been here excluded the formic and acetic acids because of some uncertainty on their molecular volume due to the formation of stable cyclic dimers
[55]. The solids and gases have been excluded in both studies in order to get experimental refractive indices only related to the polarizability.
The enthalpy of vaporization values for the 445 VOC under study come from ChemSpider [49]. The optimal MLRA obtained for the enthalpy of vaporization Hvap of these 445 VOC is as follows:
Hvap = :
· 0.4840 f n R V W + 17.23 [ F = 2554 ] (δ2020 + 17.23) (16)
· [] (ε2020) (17)
· [] (ω2020) (18)
· [] (Svap 2020) (19)
in which Svap stands for the entropy of vaporization and TBP for the boiling point expressed in kelvins.
Comments
· The predictive regressions of enthalpy values as the sum of Equations (16)-(19) applied to the 116 hydrocarbons taken alone and to the all 445 compounds of the database under study are visualized in Figure 7. It clearly appears that the intermolecular forces are better characterized for the pure hydrocarbons for which the Equation (18) and Equation (19) equal zero. Let us however note that the partial F ratios for each of these four equations are high (values between square brackets) and also that the four retained molecular characteristics in their 2020 version are quite mutually independents, as it can be seen in the following correlation matrix:
· The constant 2.25 attributed to the ε2020 definition has been chosen to provide zero values for the normal alkanes.
· The comparison between the 2016 and 2020 versions of the VOC characteristics is as follows:
(by definition) (20)
(, also from their definitions) (21)
· By contrast, ω2020 could not be assimilated to ω2020-SMT (definition recalled in Figure 6). This observation will be commented in the Discussion-Conclusion section but we already indicate our preference for ω2020.
· The constant 17.23 greatly reflects the product PV of vaporization included in the corresponding enthalpy (H = U + PV). Taking into account the important difference of molar volume in liquid and gas phases (respectively 0.1 liter in average and 24 liters) associated to a constant pressure of one atmosphere, a product value in the range 23 - 24 was expected rather than 17.23. This difference of values could be due, among others, to the predictive function of Vw including a constant, i.e. providing positive values for hypothetic compounds without any atom (see Figure 1 in [2] and note an erratum in this figure: the right F ratio value is of 211,090, not 211). This kind of theoretical drawback for a predictive model at very small data values is frequent and without practical consequences for usual data.
· According to a study of Goss and Schwarzenbach [56] on an experimental dataset of enthalpies of vaporization for more than 200 compounds, it clearly appears that intermolecular hydrogen bonding is observed not only for the well known cases of alcohols and carboxylic acids, but also for various nitro-compounds. Therefore, the Svap-2020 descriptor proposed here appears rather convincing for characterizing the entropy of vaporization, and thus in some way validates the general definitions of δ2020, ε2020 and ω2020 which together reflect the free internal energy of vaporization Uvap (), i.e. the Van der Waals forces of vaporization.
3.4. Attempt to Extend Previous Results to Compounds in Solid or Gas State at Room Temperature
The Equations (16)-(19) have been applied to another data set of 180 compounds for which interesting results have been obtained in an olfactory QSAR study as mentioned above. On the contrary to the 445 compounds dataset including only liquids at room temperature, the 180 compounds dataset includes 27 solids and 8 gases (the redundancy between these two datasets concerns about 60 compounds). As it can be seen in Figure 8, the predictive model of the enthalpy of vaporization proposed in (3.3.) is rather satisfactory not only for liquids but also for gases and solids excepted for musk xylene (coordinates 71, 62). However, it should be underlined that even though it is not specified in ChemSpider, the predicted enthalpies of vaporization values reported from ACD/labs are very probably based on the boiling point value which is not experimentally known for this compound. That could generate a cumulative fragility of its published enthalpy of vaporization. The question is still open at this stage.
4. Discussion and Conclusions
The author is greatly indebted to the Royal Society of Chemistry for its free ChemSpider database of chemical structures and physicochemical properties, including values of the named “enthalpies of vaporization” on which the present study is based.
We understand that this last expression is an abbreviation of “difference of enthalpy at the boiling point and at room temperature (usually 20˚C or 25˚C)”. It is also named “boiling enthalpy”.
The present study confirms some results on the intermolecular forces based on GLC experimentation previously published (the London and Debye forces), as stated in the Introduction of this paper. It also improves the results previously obtained on the molecular polarity strictly speaking (Keesom forces) as stated in the Results section. However, we believe that at this stage a dialog with colleagues involved in theoretical and experimental thermodynamics would be fruitful to go even further in the present field.
Some particular comments
· The descriptors of molecular polarity strictly speaking for solutes, via a GLC approach and the enthalpy of vaporization one, present some similarities but not complete.
· The first interesting convergence observed in the present study on the ω descriptor is summarized in Figure 5 and Figure 6 as well as in Equation (18): an optimal predictive model using molecular features divided by GSS (global spherical surface) rather than the original molecular features as in 2016. However, the nature of the retained molecular features differs when obtained from Hvap or GLC: the former with one of the PSA variants described in the Material and Methods section (PSA1), and the latter with 10 different molecular features including three with strong coefficients and strong partial F ratios (N3, O2 and F1) (see Figure 6).
· One hypothesis can be suggested to explain this phenomenon: some molecular proximity in the molecular aptitudes to be polar and to be proton acceptor, and a consecutive wrong rearrangement of the input values in the MMA processing of the GLC experimental data (see Figure 2). This possible mutual pollution would not be possible in the handling of Hvap data, since proton donor and proton acceptor abilities are neutralized in mutual hydrogen bonding between the molecules of a given compound. Therefore the ω values obtained from Hvap seems to be preferable.
· Another difficulty arises from the role played by the divalent sulfur compounds in our QSAR study of 2013 [38] (involvement of PSA2 rather than PSA1). Indeed, we did not found involvement of these sulfur features in the ω properties derived from GLC nor from Hvap (Equation (18), Figure 3 and Figure 6). This could perhaps be interpreted as the involvement, in the phenomenon of the human comfort OLPI (olfactory perceived intensity), of specific olfactory receptors to sulfur compounds in addition to the Van der Waals forces. It would be of course interesting to experimentally test this hypothesis using the techniques of molecular biology initiated in olfaction by Buck and Axel in 1991 [57].
· To conclude, it can be considered that the splitting of enthalpy of vaporization for liquids into δ2020, ω2020, ε2020 and [Svap-2020 + 17.23] using Equations (16)-(19) correctly reflects the Van der Vaals and the hydrogen bonding forces. It appears to be a quite robust answer entirely based on well established and easily available molecular properties. Only the three first terms are expected to be involved in some physiological phenomena, not the entropic plus the constant part.
Acknowledgements
The author reiterates its gratitude to ChemSpider for having inspired the new approach of the Van der Waals forces characterization described here. He also warmly thanks David Laffort for his strongly needed writing assistance.
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Conflicts of Interest
The authors declare no conflicts of interest regarding the publication of this paper.
Cite this paper
Laffort, P. (2020) Interest of Splitting the Enthalpies of Vaporization in Four Distinct Parts Reflecting the Van der Waals and the Hydrogen Bonding Forces. Open Journal of Physical Chemistry, 10, 117-137. https://doi.org/10.4236/ojpc.2020.102007
ReferencesLaffort, P. (2016) A Revisited Definition of the Three Solute Descriptors Related to the Van der Waals Forces in Solutions. Open Journal of Physical Chemistry, 6, 86-100. https://doi.org/10.4236/ojpc.2016.64009Laffort, P. (2018) Updated Definition of the Three Solvent Descriptors Related to the Van der Waals Forces in Solutions. Open Journal of Physical Chemistry, 8, 1-14.https://doi.org/10.4236/ojpc.2018.81001Dravnieks, A. and Laffort, P. (1972), Physicochemical Basis of Quantitative and Qualitative Odor Discrimination in Humans. In: Schneider, D., Ed., Olfaction and Taste IV, Wissens-Verlag-MBH, Stuttgart, Germany, 142-148.Eon, C. (1975) Personal Communication, Ecole Polytechnique, Route de Saclay, Palaiseau, France.Karger, B.L., Snyder, L.R. and Eon, C. (1976) An Expanded Solubility Parameter Treatment for Classification and Use of Chromatographic Solvents and Adsorbents: Parameters for Dispersion, Dipole and Hydrogen Bonding Interactions. Journal of Chromatography A, 125, 71-88. https://doi.org/10.1016/S0021-9673(00)93812-3Karger, B.L., Snyder, L.R. and Eon, C. (1978) Expanded Solubility Parameter Treatment for Classification and Use of Chromatographic Solvents and Adsorbents. Analytical Chemistry, 50, 2126-2136. https://doi.org/10.1021/ac50036a044Laffort, P. and Patte, F. (1976) Solubility Factors in Gas-Liquid Chromatography: Comparison between Two Approaches and Application to Some Biological Studies. Journal of Chromatography A, 126, 625-639.https://doi.org/10.1016/S0021-9673(01)84107-8Rohrschneider, L. (1966) Eine Methode zur Charakterisierung von Gaschromatographischen Trennflüssigkeiten. Journal of Chromatography A, 22, 6-22. https://doi.org/10.1016/S0021-9673(01)97064-5McReynolds, J. (1970) Personal Communication, IIT Research Institute, Chicago, IL.Laffort, P., Chauvin, F., Dallos, A., Callegari, P. and Valentin, D. (2005) Solvation Parameters. Part 1: Mutual Improvements of Several Approaches and Determination of Two First Sets of Optimized Values. Journal of Chromatography A, 1100, 90-107. https://doi.org/10.1016/j.chroma.2005.09.022Dallos, A., Sisak, A., Kulcsár, Z. and Kováts, E. (2000) Pair-Wise Interactions by Gas Chromatography: VII. Interaction Free Enthalpies of Solutes with Secondary Alcohol Groups. Journal of Chromatography A, 904, 211-242.https://doi.org/10.1016/S0021-9673(00)00908-0Kováts, E. and Kresz, R. (2006) Wrong Gas Liquid Partition Data by Gas Chromatography. Journal of Chromatography A, 1113, 206-219.https://doi.org/10.1016/j.chroma.2006.01.123Laffort, P. and Héricourt, P. (2009) Solvation Parameters. Part 4: What Is the Impact of Recent Chromatographic Data Update? The Open Applied Informatics Journal, 3, 12-21. https://doi.org/10.2174/1874136300903010012Cloux, R., Défayes, G., Fóti, K., Dutoit, J.C. and Kováts, E. (1993) Pair-Wise Interactions by Gas Chromatography. Part III. Synthesis of Isosteric Stationary Phases for Gas Chromatography. Synthesis, 9, 909-919. https://doi.org/10.1055/s-1993-25968Robin, P. (1970-1976) Personal. Communication. CNRS, 3 rue Michel Ange, Paris.Patte, F., Etcheto, M. and Laffort, P. (1982) Solubility Factors for 240 Solutes and 207 Stationary Phases in Gas-Liquid Chromatography. Analytical Chemistry, 54, 2239-2247. https://doi.org/10.1021/ac00250a024Laffort, P. and Héricourt, P. (2006) Solvation Parameters. 2. A Simplified Molecular Topology to Generate Easily Optimized Values. Journal of Chemical Information and Modeling, 46, 1723-1734. https://doi.org/10.1021/ci0600152Laffort, P. and Héricourt, P. (2008) Solvation Parameters of Solutes: An Attempt of Improvements in Characterization and Determination. The Open Applied Informatics Journal, 2, 22-31. https://doi.org/10.2174/1874136300802010022Laffort, P. (1968) Some New Data on the Physicochemical Determinants of the Relative Effectiveness of Odorants. In: Tanyolac, N.N., Ed., Theories of Odor and Odor Measurement, Robert College, Istanbul, 247-270.Laffort, P. (1969) A Linear Relationship between Effectiveness and Identified Molecular Characteristics, Extended to Fifty Pure Substances. In: Pfaffmann, C., Ed., Olfaction and Taste III, Rockefeller University Press, New York, 150-157Laffort, P. and Dravnieks, A. (1973) An Approach to a Physicochemical Model of Olfactory Stimulation in Vertebrates by Single Compounds. Journal of Theoretical Biology, 38, 335-345. https://doi.org/10.1016/0022-5193(73)90178-1Laffort, P., Patte, F. and Etcheto, M. (1974) Olfactory Coding on the Basis of Physicochemical Properties. Annals of the New York Academy of Sciences, 237, 193-208.https://doi.org/10.1111/j.1749-6632.1974.tb49854.xReddy, K.S., Dutoit J.C. and Kováts, E. (1992) Pair-Wise Interactions by Gas Chromatography. Part I. Interaction Free Enthalpies of Solutes with Non Associated Primary Alcohol Groups. Journal of Chromatography A, 609, 229-259.https://doi.org/10.1016/0021-9673(92)80167-SReddy, K.S., Cloux, R. and Kováts, E. (1994) Pair-Wise Interactions by Gas Chromatography. Part IV. Interaction Free Enthalpies of Solutes with Trifluoromethyl-Substituted Alkanes. Journal of Chromatography A, 673, 181-209.https://doi.org/10.1016/0021-9673(94)85037-2Défayes, G., Reddy, K.S., Dallos, A. and Kováts, E. (1995) Pair-Wise Interactions by Gas Chromatography. Part V. Interaction free Enthalpies of Solutes with Primary Chloro- and Bromo-Alkanes. Journal of Chromatography A, 699, 131-154.https://doi.org/10.1016/0021-9673(95)00023-GReddy, K.S., Cloux, R. and Kováts, E. (1995) Pair-Wise Interactions by Gas Chromatography. Part VI. Interaction Free Enthalpies of Solutes with Primary Methoxyalkane, Cyanoalkane, and Alkanethiol Groups. Journal of Chromatography A, 704, 387-436. https://doi.org/10.1016/0021-9673(95)93206-BMolinspiration (2018) Calculation of Molecular Properties and Bioactivity Score. http://www.molinspiration.com/cgi-bin/propertiesAbraham, M.H. (1993) Scales of Solute Hydrogen-Bonding: Their Construction and Application to Physicochemical and Biochemical Processes. Chemical Society Reviews, 22, 73-83. https://doi.org/10.1039/cs9932200073McReynolds, W.O. (1970) Characterization of Some Liquid Phases. Journal of Chromatographic Sciences, 8, 685-691. https://doi.org/10.1093/chromsci/8.12.685Palm, K., Luthman, K., Ungell, A.L., Strandlund, G. and Artursson, P. (1996) Correlation of Drug Absorption with Molecule Surface Properties. Journal of Pharmacological Sciences, 85, 32-39. https://doi.org/10.1021/js950285rPalm, K., Stenberg, P., Luthman, K. and Artursson, P (1997) Polar Molecular Surface Properties Predict the Intestinal Absorption of Drugs in Humans. Pharmaceutical Research, 14, 568-571. https://doi.org/10.1023/A:1012188625088Ertl, P. (2007) Polar Surface Area. In: Mannhold, R., Ed., Molecular Drug Properties, Wiley-VCH, Heidelberg, 111-126. https://doi.org/10.1002/9783527621286.ch5Ertl, P., Rohde, B. and Selzer, P. (2000) Fast Calculation of Molecular Polar Surface Area as a Sum of Fragment-Based Contributions and Its Application to the Prediction of Drug Transport Properties. Journal of Medicinal Chemistry, 43, 3714-3717.https://doi.org/10.1021/jm000942eLaffort, P. (2011) Solvation Parameters. Part 5: Physico-Chemical Interpretation of Experimental Solvent Values for Stationary Phases of Gas-Liquid Chromatography. Journal of Chromatography A, 1218, 4025-4033.https://doi.org/10.1016/j.chroma.2011.04.068Fritz, D.F. and Kováts, E. (1973) Activity Coefficient for Gas Chromatography. Influence of the Molecular Weight of the Stationary Phase on Gas Chromatographic Data. Analytical Chemistry, 45, 1175-1179. https://doi.org/10.1021/ac60329a021Défayes, G., Fritz, D.F., Gorner, T., Huber, G., De Reyff, C. and Kováts, E. (1990) Organic Solutes in Paraffin Solvents. Influence of the Size of the Solvent Molecule on Solution Data. Journal of Chromatography A, 500, 139-184.https://doi.org/10.1016/S0021-9673(00)96067-9Reddy, K.S. and Kováts, E. (1990) Dependence of Solution Data at Ideal Dilution on Solvent Molecular Size. Chromatographia, 30, 493-499.https://doi.org/10.1007/BF02269794Laffort, P. (2013) A Slightly Modified Expression of the Polar Surface Area Applied to an Olfactory Study. Open Journal of Physical Chemistry, 3, 150-156.https://doi.org/10.4236/ojpc.2013.34018Marfaing, P., Rouault, J. and Laffort, P. (1989) Effect of the Concentration and Nature of Olfactory Stimuli on the Proboscis Extension of Conditioned Honey Bees Apis mellifica Ligustica. Journal of Insect Physiology, 35, 949-955.https://doi.org/10.1016/0022-1910(89)90018-8Canard, I. (1990) Influence of the Stimulus Intensity and of the Atmosphere Ionisation on the Olfactory Learning of the Honey Bee Apis mellifica Ligustica (In French). DEA of Behavior Biology, University of Paris-Orsay, Paris, 59.Laffort, P. (1998) Olfactory Learning in the Honey Bee (In French): Some Results that Could Be Extended to Humans. In: Bertrand, G., Ed., Le Gout, Network CLUSE, Third Cross-Border Symposium, University of Bourgogne, Dijon, 12-13 September 1996, 579-584.Laffort, P. (2001) The Concept of Comfort Concentration in Olfaction from Honey Bee Experimentation. Similarities and Differences with Human Olfaction, Monell and TOSTQ Sweetness Conference Workshop, Monell Chemical Senses Centre, Philadelphia, PA, 23-24 April 2001.Patte, F., Etcheto, M., Marfaing, P. and Laffort, P. (1989) Electroantennogram Stimulus-Response Curves for 59 Odorants in the Honey-Bee Apis mellifica. Journal of Insect Physiology, 35, 667-675. https://doi.org/10.1016/0022-1910(89)90086-3Etcheto, M. (1982) Electroantennography of the Olfactory Sensitivity in the Honey-Bee (In French). University of Paris-Orsay, Paris, 139.Hill, A.V. (1913) The Combinations of Haemoglobin with Oxygen and with Carbon Monoxide. Biochemistry Journal, 7, 471-480. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1276492/ https://doi.org/10.1042/bj0070471Keil, T.A. (1982) Contacts of a Pore Tubules and Sensory Dendrites in Antennal Chemosensilla of a Silkmoth: Demonstration of a Possible Pathway for Olfactory Molecules. Tissue Cell, 14, 451-462. https://doi.org/10.1016/0040-8166(82)90039-8Laffort, P. and Patte, F. (1987) Derivation of Power Law Exponents from Olfactory Thresholds for Pure Substances. Annals of the New York Academy of Sciences, 510, 436-439. https://doi.org/10.1111/j.1749-6632.1987.tb43582.xZamora, A. (1976) An Algorithm for Finding the Smallest Set of Smallest Rings. Journal of Chemical Information and Computer Sciences, 16, 40-43.https://doi.org/10.1021/ci60005a013ChemSpider, Search and Share Chemistry. http://www.chemspider.com/Search.aspxChemaxon (2019) Molecular Surface Area Plugin 3D.https://docs.chemaxon.com/display/docs/Molecular+Surface+Area+Plugin+3DFerrara, P., Apostolakis, J. and Caflisch, A. (2002) Evaluation of a Fast Implicit Solvent Model for Molecular Dynamics Simulations. Proteins, 46, 24-33.https://doi.org/10.1002/prot.10001Partington, J.R. (1951) The Property of Liquids. In: An Advanced Treatise on Physical Chemistry, Volume 2, Longmans Green and Co, London, 17-28.Chauvin, F. and Laffort, P. (1997) Determination of a Polarizability Index by Means of Molecular Topology. Journal de Chimie Physique et de Physico-Chimie Biologique, 94, 1216-1233. https://doi.org/10.1051/jcp/1997941216Weast, R.C. (1969) CRC Handbook of Chemistry and Physics. 50th Edition, The Chemical Rubber Company, Cleveland, OH.Wikipedia (2019) Trouton’s Rule. https://en.wikipedia.org/wiki/Trouton's_ruleGoss, K.U. and Schwarzenbach, R.P. (1999) Empirical Prediction of Heats of Vaporization and Heats of Adsorption of Organic Compounds. Environmental Science and Technology, 33, 3390-3393. https://doi.org/10.1021/es980812jBuck, L. and Axel, R. (1991) A Novel Multigene Family May Encode Odorant Receptors: A Molecular Basis for Odor Recognition. Cell, 65, 175-187.https://doi.org/10.1016/0092-8674(91)90418-X
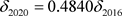 (
( by definition) (20)
by definition) (20) (
( , also from their definitions) (21)
, also from their definitions) (21)