Synthesis and Spectroscopic Properties of Ferrocenyl Derivative Containing Donor and Acceptor Groups ()
1. Introduction
Large second-order hyperpolarizabilities are known to be associated with the structure of molecules that have large differences in dipole moments between ground- and excited-states. A typical NLO (nonlinear optical) material consists of an electron-accepting group, a bridge, and an electron-donating group. The bridge is conjugated, thus allowing for good communication between the donor and the acceptor moieties. This sort of materials is useful in optical frequency conversion and optical data processing, and is expected to play a central role in the next generation of optoelectronics [1] [2] .
Although investigations were initially focused on purely inorganic system, a wide variety of organic NLO materials have now been discovered and have been vigorously investigated [3] [4] . In addition, since the report in 1987 by Green et al. that a ferrocenyl derivative A (Scheme 1) had an excellent SHG efficiency (62 times of the urea standard) [5] , much interest has been devoted to organometallic compounds possessing this property [6] [7] .
It has been pointed out that organometallic structures are intriguing candidates for study as NLO chromophores by virtue of their low-energy, yet sometimes intense, electronic transitions. Especially, metallocene derivatives have been the most thoroughly investigated compounds to date. In these compounds, metallocenyl groups are utilized as an effective electron donor. Indeed, it was found that the dipole projections of the β tensors of ferrocene complexes are comparable to those of methoxyphenyl systems with similar acceptors [8] . We have become interested in exploring the utility of ferrocenyl group as the conjugating bridge because both aromatic cyclopentadienyl rings in ferrocene interact with iron atom through their π-electron systems [9] . Previously Marder and co-workers observed that replacement of a cyclopentadienyl ring in complex A with a more electron donating pentamethylcyclopentadienyl ring leads to increased optical nonlinearity [8] . On the basis of a similar line of reasoning, we envisioned that incorporating an electron-rich substituent at one of the cyclopentadienyl rings would enhance the NLO properties.
We report herein the facile synthesis of a novel ferrocenyl derivative 1 (Scheme 2) bearing conjugated electron donating and accepting substituent in 1,1’-positions starting from ferrocenyl dicarbaldehyde 2. Syntheses of such asymmetrical disubstituted ferrocenes have become the focus of some attention [10] .
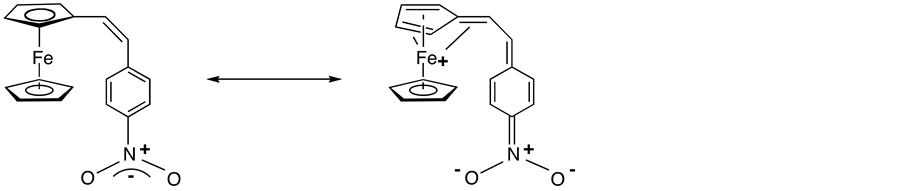
Scheme 1. Two canonical representation of A.
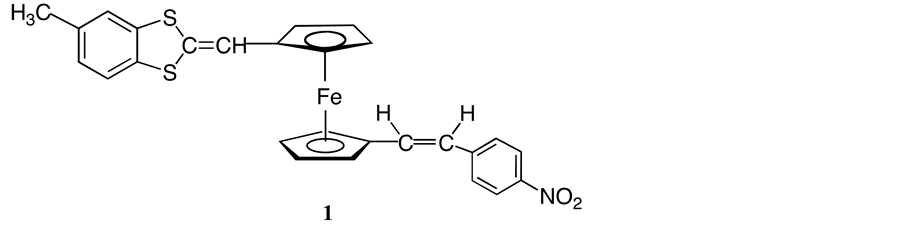
Scheme 2. Target compound 1.
2. Experimental
2.1. General Considerations
Unless otherwise noted, all reactions were carried out under anaerobic and anhydrous conditions using Ar or N2 and conventional Schlenk techniques using the general methods. Solvents were dried and purified in the usual manner, and stored under an atmosphere of Ar. Commercially available chemicals were used as such without any further purification. Infrared spectra (IR) were determined on a Perkin-Elmer 1600 series spectrometer using KBr disks prepared under inert atmosphere. NMR spectra were measured using a JEOL JNMEX270 SPECTROMETER at 270 MHz (1H) and 67.5 MHz (13C). 1H- and 13C-NMR chemical shifts were referenced to trimethylsilane (TMS). UV-vis absorption spectra were recorded on a JASCO model V-550 spectrophotometer. The determination of the second harmonic intensity of powder samples was made relative to that of powdered mNA (meta-nitroaniline) using an SHEW (second-harmonic generation with the evanescent wave method). The details of the experimental arrangement were described elsewhere [11] . Literature methods were used to prepare ferrocene-1,1’-dicarbaldehyde 2 [12] and Bu3P=CH-4-C6H4NO2 (7) [13] .
2.2. Synthesis of 2-(Dimethoxyphosphoryl)-1,3-(5-Methylbenzo)Dithiol (5)
This compound was prepared starting from toluene-3,4-dithiol by using known procedures (58% overall yield) [14] , [15] , [16] . IR (KBr) δ (C-H) 1436, 1390, ν (P=O) 1251, ν (P-O) 1040 cm−1; 1H-NMR (CDCl3) δ = 7.03 - 7.09 and 6.82 (m, aromatic), 4.87 (d, 2JPH = 5.3 Hz, 1H, S2CH), 3.79 (d, 3JPH = 10.6 Hz, 6H, POCH3), 2.26 (s, 3H, C6H3CH3).
2.3. Synthesis of 1-{{[1,3-(5-Methylbenzo)Dithiol]-2-Ylidene}Methyl}-1’-Formylferrocene (6)
A solution of 5 (1.66 g, 6.01 mmol) in THF (80 mL) was treated with n-BuLi (4.2 mL, 1.6 M in hexane, 6.61 mmol) at −78˚C. Then a cooled (−78˚C) solution of 2 (7.26 g, 30.0 mmol) in 200 mL of THF was added dropwise. The reaction mixture was stirred at this temperature for 90 min before being allowed to warm slowly to room temperature and stirred overnight. The solvent was evaporated to dryness under reduced pressure. The residue was transferred to a silica gel column and chromatographed with dichlorometane-hexane (1:3) to give compound 6 (0.27 g, 12%). IR (KBr) ν (C-H, aromatic) 3420, ν (C-H, CHO) 2921, ν (C=O) 1681, ν (C=C) 1578 cm−1. 1H-NMR (C6D6) δ = 9.89 (s, 1H, C5H4CHO), 6.55 - 6.80 (m, 3H, aromatic), 5.89 (s, 1H, S2CCHC5H4), 4.56 and 4.14 (s, 2H; s, 2H, C5H4CHO), 4.32 and 3.97 (s, 2H; s, 2H, S2CCHC5H4), 1.88 (s, 3H, C6H3CH3); 13C-NMR δ = 192.1 (C5H4CHO), 136.9 (S2CCHC5H4), 136.0, 135.7, 131.0, and 121.0 - 127.1 (aromatic), 110.2 (S2CCHC5H4), 85.1, 69.9, and 68.7 (S2CCHC5H4), 80.8, 74.0, and 70.6 (C5H4CHO), 20.7 (C6H3CH3).
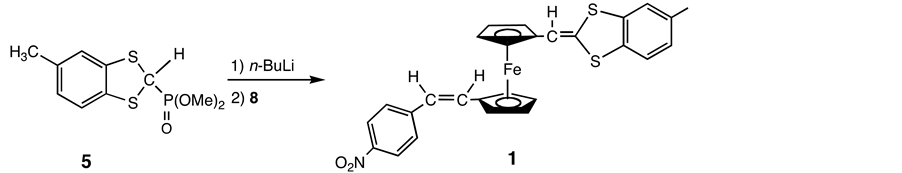
2.4. Synthesis of 1-{{[1,3-(5-Methylbenzo)Dithiol]-2-Ylidene}Methyl}-1’-[2-(p-Nitrophenyl)-(E)-Ethenyl]Ferrocene (1)
A solution of 7 (0.39 g, 1.04 mmol) in toluene (20 mL) was treated with n-BuLi (0.73 mL, 1.6 M in hexane, 1.14 mmol) under Ar at −78˚C. The solution was stirred for 60 min at −78˚C, then allowed to warm slowly to room temperature. The reaction mixture was stirred at this temperature for 120 min. To a cooled (0˚C) solution of 0.27 g (0.69 mmol) of 6 in 20 mL of diethyl ether was added the above solution and the resulting mixture was allowed to warm slowly to room temperature, then stirred overnight. The solvent was removed under reduced pressure and purification using preparative TLC on silica gel with dichloromethane-hexane (1:1) yielded 1 (0.010 g, 2.8%) as a purple powder. IR (KBr) ν (C=C) 1626 and 1591, ν (NO2) 1509 and 1339 cm−1: 1H-NMR (C6D6) δ = 7.76 (d, J = 8.9 Hz, 2H, CHCHNO2), 7.08 (d, J = 8.9 Hz, 2H, CHCHNO2), 6.77 - 6.82 and 6.55 - 6.66 (m, 3H, aromatic), 6.26 (d, J = 11.9 Hz, 1H, C5H4CH=CH), 6.20 (d, J = 11.9 Hz, 1H, C5H4CH=CH), 5.95 (s, 1H, S2CCHC5H4), 4.35 and 4.06 (s, 2H; s, 2H, S2CCHC5H4), 4.12 and 4.01 (s, 2H; s, 2H, CHCHC5H4), 1.96 (s, 3H, C6H3CH3); 13C-NMR δ = 148.0 (CNO2), 146.0 (CCHCHCNO2), 138.0 (S2CCHC5H4), 137.0 (CS2CCH), 133.4 (H3CCCH), 132.9 (C5H4CH=CH), 130.6 (CHCHCCNO2), 128.4, 123.8 and 122.2 (aromatic CH, methylbenzodithiol ring), 127.0 (C5H4CH=CH), 124.7 (CHCNO2), 112.6 (S2CCHC5H4), 85.8 (S2CCHC), 82.5 (CCH=CH), 72.2 (CHCHCCH=CH), 71.7 (S2CCHCCHCH), 71.1 (CHCCH=CH), 70.3 (S2CCHCCH), 22.0 (C6H3CH3). Anal. Found: C, 62.61; H, 3.97; N, 2.74. Calcd for C27H21NO2S2Fe: C, 63.41; H, 4.14; N, 2.74.
2.5. Synthesis of 1-[2-(p-Nitrophenyl)-(E)-Ethenyl]-1’-Formylferrocene (8)
A solution of 7 (3.09 g, 8.27 mmol) in toluene (150 mL) was treated with n-BuLi (5.8 mL, 1.6 M in hexane, 9.1 mmol) under Ar at −78˚C. The solution was stirred for 60 min at −78˚C, then allowed to warm slowly to room temperature. The reaction mixture was stirred at this temperature for 120 min. To a cooled (0˚C) solution of 3.00 g (12.4 mmol) of 2 in 150 mL of diethyl ether was added the above solution and the resulting mixture was allowed to warm slowly to room temperature, then stirred overnight. The solvent was removed under reduced pressure and the residue was chromatographed on a silica gel eluted with toluene/hexane (10:1), affording 1.43 g (48%) of 8 as a purple powder. IR (KBr) ν (C=O) 1678, ν (C=C) 1631 and 1590, ν (NO2) 1508 and 1336 cm−1: 1H NMR (C6D6) δ = 9.72 (s, 1H, C5H4CHO), 7.90 (d, J = 8.9 Hz, 2H, CHCHNO2), 6.94 (d, J = 8.9 Hz, 2H, CHCHNO2), 6.52 (d, J = 16.2 Hz, 1H, C5H4CH=CH), 6.37(d, J = 16.2 Hz, 1H, C5H4CH=CH), 4.47 and 4.03 (s, 2H; s, 2H, C5H4CHO), 4.19 and 3.97 (s, 2H; s, 2H, CHCHC5H4); 13C-NMR δ = 192.2 (C5H4CHO), 147.2 (CNO2), 143.5 (CCHCHCNO2), 130.4 (C5H4CH=CH), 126.4 (CHCHCCNO2), 125.8 (C5H4CH=CH), 124.3 (CHCNO2), 83.9 (CCH=CH), 81.0 (CHOCCH), 73.8 (CHOCCHCH), 71.1 (CH=CHCCHCH), 70.9 (CHOCCH), 68.7 (CH=CHCCH).
2.6. Reaction of 5 with 8
A solution of 5 (1.66 g, 6.01 mmol) in THF (100 mL) was treated with n-BuLi (4.2 mL, 1.6 M in hexane, 6.61 mmol) at −78˚C under Ar. The solution was stirred for 90 min at −78˚C. Then a cooled (−78˚C) solution of 8 (1.81 g, 5.01 mmol) in 100 mL of THF was added dropwise. The reaction mixture was stirred at this temperature for further 120 min before being allowed to warm slowly to room temperature and stirred overnight. The solvent was evaporated to dryness under reduced pressure. Then the resulting residue was transferred to a silica gel column and chromatographed with hexane-ethyl acetate. Compound 1 was further purified using preparative TLC (0.050 g, 2.0%).
3. Results and Discussion
We chose the sulfur-based substituent, which can be derived from toluene-3,4-dithiol, as the electron-donating group since Togni and co-workers had found that the analogous CH=CS2C6H4 group could pour a significant amount of electron density on the ferrocene moiety [17] . They reported that analysis of the cyclic voltammograms exhibited this substituent makes the electron removal easier with respect to ferrocene by about 100 mV. As the electron-accepting group we chose nitrobenzene conjugating moiety, which enhances the molecular hyperpolarisabilities of the compound [5] .
The synthetic route commenced with known ferrocenyl dicarbaldehyde 2. This compound is readily available from the reaction of 1,1’-dilithioferrocene with DMF [12] . Compound 1 would be synthesized from the successive reactions of 2 with the appropriate Wittig- and Wittig-Horner reagents (Scheme 3).
The donating group was accessible in four steps from toluene-3,4-dithiol, which was initially converted into S,S’-methylene derivative 3 [14] . Treatment of 3 with trityl fluoroborate in CH3CN afforded 1,3-dithiolylium tetrafluoroborate 4 [15] , which reacted with trimethyl phosphite in the presence of one molar amount of sodium iodide [16] . Resulting 2-methoxyphosphinyl-1,3-dithiol 5 was converted into the phosphonate ylide by n-BuLi, and treatment of the Wittig- Horner reagent with one molar amount of the ferrocenyl dicarbaldehyde 2 in THF gave mono- and di-substituted compounds respectively [17] .
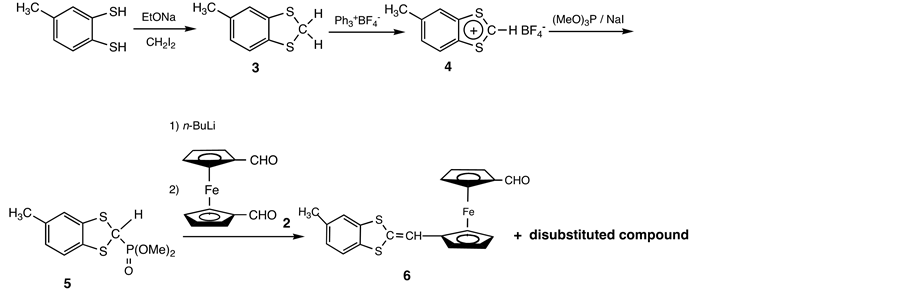
Scheme 3. Synthesis of 6.
Thus under these conditions, the major product was found to be a disubstituted compound and the desired monosubstituted aldehyde 6 was obtained in only 2% yield on chromatographing the reaction mixture. Employment of substantial excess (5-fold) of 2 led to improved result (12%) but the major product was again the disubstituted compound. It seemed likely that the electron-do- nating group that was initially introduced into one of the Cp rings strongly activates the remaining formyl group. The reaction of the monosubstituted aldehyde 6 with the ylide Bu3P=CH-4-C6H4-NO2, which was prepared from 4-nitrobenzyl chloride [13] , afforded a complex mixture. Purification of the mixture via conventional chromatography gave compound 1, though in low yield (Scheme 4).
During the purification, we observed the formation of side-products. Hence, the oxidative decomposition of 1 in solvents under air might have taken place. It has been pointed out that ferrocenyl derivatives sometimes decompose in a dioxygen-containing solution to unidentified, insoluble oxidized products [18] . The resulting compound 1 displays reasonable air and thermal stabilities in the solid state. Complex 1 was fully characterized by spectroscopic methods as well as by elemental analyses (vide infra).
An alternative route, in which the electron-accepting group is introduced to 2 prior to the introduction of the electron-donating group, was also explored. Thus 2 reacted with the ylide Bu3P=CH-4-C6H4-NO2 (7) to give the corresponding derivative 8 (Scheme 5).
Preliminary experiments revealed that the relative E:Z ratios and the yields were found to depend upon the solvent used. The highest proportions of the trans-olefin were obtained in diethyl ether-toluene mixtures. Although the highest yield was obtained in DMF-toluene mixtures, the selectivity of the products was low in this solvent. In contrast to the above reaction between 2 and 5, the present reaction proceeded well and gave only a trace of the disubstituted compound. The two isomers (E and Z) are readily distinguished by 1H-NMR
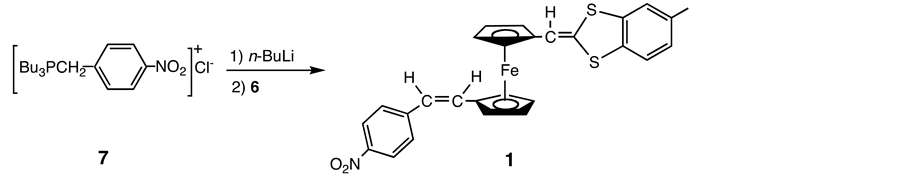
Scheme 4. Synthesis of 1.
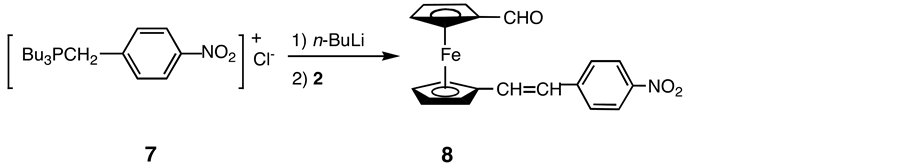
Scheme 5. Synthesis of 8.
spectroscopy, where distinctive patterns appear for each olefin protons. The E-isomer always exhibits an AB quartet, with JHH = 16 Hz, downfield to the pair of doublets (JHH = 12 Hz) exhibited by the Z-isomer [19] [20] .
Treatment of the E-isomer of 8 with the phosphonate ylide 5 in THF led to a complex mixture. A crude separation was effected by a silica gel column and isolated 1 was further purified by preparative thin-layer chromatography (TLC) (Scheme 6).
1H-NMR analysis of 1 revealed that it is a Z-isomer, as is judged from the 1H-NMR coupling constant of the CH=CH ethylene bridge (J = 11.9 Hz) as mentioned above. We have not isolated the E-isomer from a product mixture, which suggests that E/Z isomerization of this ethylene bridge occurred during the reaction since the starting complex 8 was E-isomer. We believe that the isomerization reaction occurred in the purification step. In support of this, Green and co-workers observed that E-isomer of A was converted into Z-isomer by hydrochloric acid and they postulated that a Z/E equilibration of complex A catalyzed by acidic residues in a column [19] . Use of an alumina column was found to be somewhat less effective for purification of the product. Since such isomerization did not take place in the reaction between 2 and the ylide 7, it may be envisioned that the intermediate ion can be stabilized by the electron-donating group which is attached to one of the cyclopentadienyl ring in 1 (Scheme 7).
It has been noted that there is a correlation between the solvatochromism of a molecule and the magnitude of the molecular second-order polarizability (β) [21] . Thus UV-vis absorption spectra in cyclohexane, in dichloromethane, and in DMF were recorded for compound 1 (Table 1).
Scheme 6. Reaction of 8 with 5.
![]()
Table 1. Absorption spectral data for complex 1 and Aa.
a [5] .

Scheme 7. Isomerization 1E to 1Z catalyzed by acid.
In nonpolar cyclohexane and in moderately polar dichloromethane, there were four absorptions at λmax = 475, 326, 233, and 207 nm, and at λmax = 488, 328, 243, and 234 nm respectively. On the other hand, in polar DMF there were only two absorptions in this region: one at λmax = 326 nm; the second at λmax = 502 nm. These observations are compatible with those of complex A. Green and co-workers reported that A showed three absorptions in heptane, whereas in DMF it showed only two absorptions [5] . Marder and co-workers reported that the UV-vis absorption spectra of acceptor substituted ferrocenylethylenes typically have two strong bands that are attributed to charge-transfer transitions [8] . In our case, the spectra of 1 measured in DMF indicated that incorporation of CH=CS2C6H3(CH3) group led to 10- and 14-nm shifts to the lower and higher energy bands, respectively, compared with complex A. Marder observed that pentamethyl substitution of one cyclopentadienyl ring of A led to the red shift of the both bands [8] . Interestingly the inverse shift of the higher energy band, which was assigned to a ligand π toπ* transition with some metal character [8] , was found in 1.
As shown in Table 1, the absorptions are all strong with ε values ranging from about 2000 to 60,000 M−1∙cm−1. These high extinction coefficients provide evidence for extensive charge-transfer interaction [22] . Some shifts in the positions of the absorption bands occurred when the solvent polarity is changed and these differences were considerably large; compound 1 exhibits a positive solvatochromic behavior.
The second-order NLO property of compound 1 was assessed using an SHEW (second-harmonic generation with the evanescent wave) technique [11] . This technique has significant advantages: the SHEW power is not sensitive to the birefringent phase-matching condition of a sample and the d coefficient can be obtained using a powder sample. Based on the SHEW power as a function of the incidence angle of the fundamental wave, the effective d value (deff) relative to a mNA (meta-nitroaniline) standard was measured at 1.06 mm. Although d is a tensor defined in the crystalline coordinates, the obtained deff was directionally averaged scalar value because the powder crystals oriented in random directions. deff relative to the standard is approximately counted as the relative value of the largest component of d tensor. The SHEW technique has been shown to provide more appropriate results for common organic SHG materials than the powder test usually used [11] .
d for the compound 1 was roughly estimated to be 1 pm/V using d33 (mNA) = 15 pm/V [23] . Therefore the efficiency is not so high. However the results establish the synthetic route to prepare a ferrocene derivative which has a donor and an acceptor groups in 1,1’-positions. This approach may offer a more effective means of preparing ferrocene derivatives that have an excellent SHG efficiency. We are searching for more effective donor groups.
4. Conclusion
Successive reactions of ferrocene-1,1’-dicarbaldehyde with Wittig and Wittig-Horner reagents, Bu3P=CHC6H4NO2 and [(MeO)2OPCS2C6H3(CH3)]-, yielded formation of 1. Complex 1 was stable in the solid state but deteriorated gradually in solution. This made it difficult to obtain 1 with good yield. The UV/vis spectra of 1 showed considerable solvatochromism. 1 was tested for non-linear optical properties, while the result was negative.
Acknowledgements
We thank Dr. M. Tanaka of Tokyo Institute of Technology for the elemental analysis. We also gratefully acknowledge several useful discussions with Prof. Yoshio Taniguch.