Pharmacokinetic Evaluation of Chitosan-Succinyl-Prednisolone Conjugate Microparticles as a Colonic Delivery System: Comparison with Enteric-Coated Conjugate Microparticles ()
1. Introduction
Recently, inflammatory bowel disease (IBD), represented by ulcerative colitis (UC) and Crohn’s disease (CD), has become a serious health problem for many developed countries. IBD is said to be induced by various factors such as genetic background, change in intestinal environment associated with enteric flora and disorder of intestinal immune system [1] -[3] . However, as the root cause has not been elucidated clearly yet, its treatment is mainly based on a symptomatic therapy, not a definitive therapy. Although new agents such as antibodies against proinflammatory cytokines have been developed as new excellent drugs, they are very costly and not easy to use due to immune system-related side effects [4] [5] ; therefore, conventional drugs such as 5-ASA-related agents and glucocorticoids are still important clinically [6] [7] .
Prednisolone (PD) is often used for the treatment of IBD because it can function effectively and promptly even to the severe diseased states [8] [9] . However, as PD causes severe toxic side effect considerably in a prolonged use or high dose, its use is generally limited [10] [11] . It is important for enhancement of efficacy and reduction of toxic side effect to deliver an anti-inflammatory drug to the diseased site specifically. As to 5-ASA, sulfasalazine, Pentasa and Asacol, being commercially available, can carry 5-ASA well to the lower intestine including a colonic area; they can promote efficacy and improve toxicity [12] . As to glucocorticoids, various targeted delivery systems have been developed based on a similar concept [13] [14] . In particular, for UC, as the diseased area located in the lower intestine, the techniques such as delayed release or enteric-coating have been attempted to achieve effective targeting [15] -[18] , though the systems related to glucocorticoids have not been commercially available. Microor nano-particles and prodrugs for glucocorticoids have been reported to exhibit enhanced efficacy and reduced toxicities, which are based on the localization of the drug at the diseased site and the reduction of the systemic drug level [19] -[21] . Thus, small size particles (submicron-micron order size) and prolonged release are considered to be important in order to improve the drug function in the treatment of UC [19] [22] .
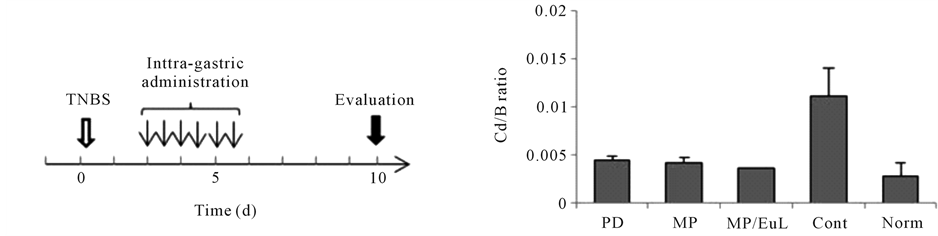
From such knowledge, PD-loaded microparticles have been developed using chitosan-suscinyl-predinisolone conjugate and enteric-coating polymer, Eudragit [23] [24] . Before now, Eudragit L-coated conjuagte microparticles (MP/EuL) were evaluated for pharmacological and pharmacokinetic features [25] [26] . The results in efficacy and toxicity are summarized in Figure 1 [26] . MP/EuL showed the most beneficial results in enhanced efficacy and reduced toxicity, which were found to be based on their pharmacokinetic features. However, conjugate microparticles (MP) themselves were found to exhibit excellent results nearly as well as MP/EuL. Namely, a question whether enteric-coating is necessary or not has remained to be solved. In this study, MP were investigated for their pharmacokinetic features, and evaluated by comparison with MP/EuL reported before [25] . As many words or phrases were abbreviated in the paper, their abbreviations were listed in Table 1.
2. Materials and Methods
2.1. Materials
Prednisolone (PD), N-hydroxysuccinimide (NHS) and chitosan (Ch; viscosity grade 1000 (5 g/L, 20˚C)) were purchased from Wako Pure Chemical Industries, Ltd. (Osaka, Japan). Prednisolone 21-hemisuccinate (SP) sodium salt (SP-Na), 2,4,6-trinitrobenzenesulfonic acid (TNBS) and 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDC) were obtained from Sigma-Aldrich (St. Louis, MO, USA). Sorbitan sesquioleate (SO-15) was obtained from Nikko Chemicals Co., Ltd. (Tokyo, Japan). All other chemicals were of reagent grade.
2.2. Animals
Male Wistar rats (7 weeks old; weighing 200 - 210 g) were purchased from Tokyo Laboratory Animals Science Co. Ltd. (Tokyo, Japan). They were bred on the breeding diet MF supplied by Oriental Yeast, Co., Ltd. (Tokyo,
 (a)
(a) (b)
(b)
Figure 1. Evaluation of anti-inflammatory effect and toxic side effect after intra-gastric administration to rats with TNBSinduced colitis at the dose of 5 mg PD eq./kg × 6 (12 h intervals). The results are based on the previous report [26] . (a) administration schedule (PD: PD alone; MP: Conjugate mcroparticles, MP/EuL: Eudragit L-coated MP, Cont: Comtrol, Norm: Normal); (b) Cd (Distal colon weight)/B (Body weight) ratio; (c) MPO activity; (d) T (Thymus weight)/B (Body weight) ratio. The results are expressed as the mean ± S.E. (n = 3 - 4).
Japan) with water ad libitum at 23˚C ± 1˚C and relative humidity of 60% ± 5%. They were used for the experiments a few days after purchase. The experimental protocol was approved by the Committee on Animal Research of Hoshi University, Japan. The animal experiments were performed in compliance with the Guiding Principles for the Care and Use of Laboratory Animals, Hoshi University, Japan.
2.3. Preparation of Conjugate Microparticles (MP)
MP were produced by referring to the previous reports [25] [26] , though the method was modified to some extent. In the first place, the conjugate between Ch and SP was prepared by the carbodiimide coupling of amino groups of Ch and the carboxyl group of SP. Briefly, Ch (120 mg) was dispersed in 85 ml of water, and dissolved by adjusting the solvent pH to 3 with a 1 M HCl aqueous solution. After SP-Na (40 mg) was added to the Ch solution, the mixture pH was adjusted to pH 5.5 with a 1 M NaOH aqueous solution and EDC (400 mg) and NHS (240 mg) were added. The resultant mixture was stirred for 3 h under ice cooling and subsequently for 21 h at room temperature. The product was precipitated with addition of a four-fold volume of acetone, and washed with a mixture of acetone and water (4:1, v/v). The product was lyophilized to obtain the conjugate powder.
Conjugate microparticles (MP) were prepared by emulsification and subsequent evaporation. The obtained conjugate (150 mg) were dissolved in a 1% (v/v) acetic acid aqueous solution (30 ml), and emulsified in 450 ml of liquid paraffin containing 1% (w/v) SO-15 at 1200 rpm at 40˚C. The emulsion was sonicated at 45˚C under 28 kH for 10 min, and the acetic acid and water were evaporated under reduced pressure while the mixture was being stirred at 400 rpm at 40˚C for 12 h. Then, the mixture was centrifuged at 3000 rpm for 5 min to precipitate the product. The precipitate was washed with n-hexane, and dried in a desiccator to obtain MP.
2.4. Particle Characteristics and in Vitro Drug Release of MP
The particle size and shape of MP were observed using an OLYMPUS BX51 optical microscope (OLYMPUS Corp.; Tokyo, Japan). The Green diameter (Feret diameter) was measured for 100 microparticles chosen at random. At the same time, their particle shape was observed. The drug content of MP was analyzed by referring to the previous study. Briefly, the MP sample (2 mg) was suspended in a 0.1 M NaOH aqueous solution (10 ml), incubated for 10 min at 45˚C and centrifuged at 3000 rpm for 5 min. The UV absorbance of the supernatant was measured at 246 nm using a Beckman DU 640 spectrophotometer to determine the PD concentration.
The in vitro release from MP was investigated using artificial gastric fluid (JP16 1st fluid (pH 1.2)) and artificial intestinal medium (JP16 2nd fluid (pH 6.8)). Namely, MP were put in 5 ml of JP16 1st and 2nd fluids at a concentration of 50 μg PD eq./ml. Each mixture was incubated by horizontal shaking at 100 rpm in a water bath warmed at 37˚C. At 1 and 6 h, the mixture was centrifuged at 3000 rpm for 5 min, and the aliquot sample (100 μl) was withdrawn and stored in a freezer at −20˚C until analysis. After each sampling, the incubation mixture was gently stirred, and the incubation was continued. The incubation time was defined as the time for the mixture to be immersed in the water bath at 37˚C. To each sample, a 3-fold volume by addition of the HPLC mobile phase was added, and the resultant mixture was analyzed by HPLC to determine the amount of released PD.
2.5. Animal Experiments
Animal models with ulcerative colitis were produced using instillation of TNBS to the colonic parts of rats [19] [20] [22] . Namely, after rats were fasted for 48 h, 0.25 mL of 50% (v/v) aqueous ethanol containing TNBS (20 mg) was instilled to each rat into the colonic part 7 cm from the anus through a catheter. Three days after TNBS treatment, the rats, which weighed 80% - 100% of that immediately before TNBS instillation, were selected as an ulcerative colitis animal model. Then, the rats were fasted for 24 h. MP were administered intragastrically at a dose of 5 mg PD eq./kg as a suspension in saline (1.5 ml). Immediately before and 0.5, 1, 2, 4, and 7 h after administration, blood samples (0.3 ml) were withdrawn via the jugular vein under light anesthesia with ethyl ether inhalation. Plasma was obtained after centrifugation of the blood at 3000 rpm for 10 min. To the plasma (100 μl), 100 μl of saturated NaCl aqueous solution, 100 μl of 6% (w/v) phosphoric acid and 4 ml of the mixture of t-tributyl methyl ether and pentane (3:2, v/v) were added, and the resultant mixture was shaken vigorously. After the mixture was centrifuged at 3000 rpm for 10 min, 3 ml of the organic phase was taken, and dried under nitrogen gas. The resultant residue was dissolved in the HPLC mobile phase, and analyzed by HPLC.
One h after blood sampling at 7 h, the animals were sacrificed by excessive inhalation of ethyl ether immediately. Their stomach (ST), upper half of the small intestine (upper small intestine or proximal small intestine (PI)), lower half of the small intestine (lower small intestine or distal small intestine (DI)), cecum (CE), upper one-third of the colon (proximal colon (PC)) and lower two-thirds of colon (distal colon (DC)) were excised. The content in each tissue was collected by washing with phosphate-buffered saline, pH 7.4 (PBS), homogenized using a glass homogenizer with a Teflon pestle, and diluted with PBS into 4 ml for ST, PI, DI and PC, and into 8 ml for CE and DC. First, the amount of free PD in the content suspension was examined as follow. Each content suspension (100 μl) was treated in the same manner as in plasma. That is, 100 μl of saturated NaCl aqueous solution, 100 μl of 6% (w/v) phosphoric acid and 4 ml of the mixture of t-tributyl methyl ether and pentane (3:2, v/v) were added, and the resultant mixture was shaken vigorously. Then, 3 ml of the organic phase was dried under nitrogen gas at room temperature. The resultant residue was dissolved in the HPLC mobile phase, and analyzed by HPLC.
In addition, the amount of total (free plus incorporated) PD in the content suspension was determined as follows. To 100 μl of the content suspension, 20 μl of 0.1 M NaOH aqueous solution was added, and kept at 45˚C for 10 min to hydrolyze the ester. Then, extraction of PD and preparation of the sample for the HPLC assay were performed as stated above. The obtained sample solution was analyzed by HPLC to determine the total amount of PD in the content suspension.
2.6. HPLC Assay
HPLC analysis of each sample was performed at room temperature using the following apparatuses and conditions, which were referred to the reports on another macromolecular prodrug of PD [27] . A Shimadzu LC-6AD pump was used with a Shimadzu SPD-10AV VP UV-VIS detector, which was set at a wavelength of 246 nm and connected with a Shimadzu C-R7A Chromatopac. A YMC Pack ODS-AM column (6 mm inner diameter × 150 mm length; YMC Co., Ltd., Kyoto, Japan) was used as the analytical column. A 26% (v/v) 2-propanol aqueous solution containing 0.1% (v/v) trifluoroacetic acid was used as the mobile phase. The HPLC assay was performed at room temperature, in which the flow rate was set at 1 ml/min and the injection volume was 20 μl. The PD concentration was determined by the absolute calibration curve method.
2.7. Statistical Analysis
Statistical analyses were performed with one-way ANOVA followed by Tukey-Kramer’s test, and the significant difference was set as p < 0.05.
3. Results and Discussion
3.1. In Vitro Characteristics
MP were prepared based on the previous method, but modified to some extent; because, in the previous production of MP, the conjugation reaction time was fairly long and the formation of microparticles required high temperature and long time. In the present study, NHS was added in the EDC conjugation of SP with Ch. The drug content of the conjugate tended to be a little greater (data not shown), and the conjugation reaction time could be shortened to 24 h. For the preparation of MP, acetic acid and water of the aqueous droplets in the emulsion could be removed for a short time (12 h) at low temperature (40˚C) by reduced pressure. The obtained microparticles (MP) are shown in Figure 2. The shape was almost spherical and the size was distributed at the range of 1 - 4 μm. The particle diameter and drug content are shown in Table 2. Their values were almost equivalent to those of the inside microparticles of MP/EuL reported before [25] [26] .
Furthermore, MP were examined for the in vitro release. The results are shown in Figure 3. The drug release patterns were similar to those of the inside microparticles of the previous MP/EuL. Although a small initial release was observed, it was considered to be due to the co-existent of PD relieved in particle formation.
PD was released gradually in the artificial intestinal fluid (pH 6.8), while very slowly in the artificial gastric fluid (pH 1.2). Therefore, although MP were produced by the modified preparation method, they were considered to have in vitro features quivalent to those of the inside microparticles of MP/EuL reported before [25] [26] .
3.2. Plasma Concentration-Time Profiles
The plasma concentration-time profile after intragastric administration of MP is described in Figure 4, in which it (closed square) was compared with those for previously-reported PD alone and MP/EuL (open triangle and

Figure 2. Optical micrographs of MP (Magnification: ×2500).

Figure 3. In vitro release profiles of PD in the incubation of MP in the JP16 1st fluid (pH 1.2) and 2nd fluid (pH 6.8). The results are expressed as the mean ± S.D. (n = 3). As the S.D. value was smaller than the symbol size, it could not be seen.

Table 2. Particle size and drug content of each preparation.
Particle diameter (n = 100); drug contentent (n = 3). aThe data of MP/EuL were based on the previous papers [25] [26] .
circle, respectively). The plasma concentration was suppressed extensively in MP as well as MP/EuL when compared with that in PD alone. Since the systemic absorption of PD decreased greatly in MP and MP/EuL, the toxic side effect represented by T/B reduction was considered to be reduced in MP as well as MP/EuL. However, the magnified figure (Figure 4(b)) showed that the PD levels increased slowly in MP, while it did not in MP/EuL. This indicated that PD was absorbed slowly for a prolonged period in MP, suggesting that MP should be inferior to MP/EuL from a viewpoint of the reduction of toxicity.
The pharmacokinetic parameters were calculated using a program MULTI [28] , and are shown in Table 3. The AUC values of MP and MP/EuL were suppressed at 1/14 and 1/70 of that of PD alone, respectively; they were significantly different from that of PD alone (p < 0.01). This was considered to lead to the reduction of

Figure 4. Plasma concentration-time profiles (a) after intra-gastric administration at 5 mg PD eq./kg to rats with TNBS-induced colitis. (b) Magnified figure for the square part. The data of MP/EuL and PD were based on the previous report [25] . The results are expressed as the mean ± S.E. (n = 3).
Table 3. Pharmacokinetic parameters of each preparation after intragastric administration at 5 mg PD eq./kg in rats with TNBS-induced colitis.
aThe data of MP/EuL and PD were based on the previous report [25] . The results are expressed as the mean ± S.E. (n = 3). *1p < 0.05 vs. PD alone, *2p < 0.05 vs. MP/EuL, #1p < 0.01 vs. PD alone, #2p < 0.01 vs. MP/EuL (Tukey-Kramer’s test).
toxic side effect. Also, the Cmax values were lowered in MP and MP/EuL. On the other hand, Tmax, MRT and VRT were significantly different between MP and MP/EuL. This was due to the gradual increase of the plasma levels in MP as shown in Figure 4(b). This plasma concentration pattern of MP was unsuitable for suppression of toxic side effects.
3.3. Gastrointestinal Drug Distribution
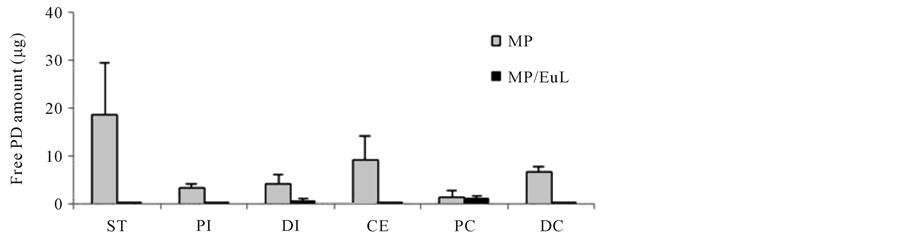
Gastrointestinal distribution was examined 8 h after intragastric administration of MP. The results are shown in Figure 5. The distribution profiles were compared with those of the previously-reported MP/EuL [25] . After the dosing of MP, free PD was observed in the tracts from ST to DC, especially, at a fairly high level in the ST. On the other hand, after the administration of MP/EuL, free PD was hardly detected throughout the gastrointestinal tract, except for the PC site. However, the total PD was observed to be distributed mainly in the DI and CE and colonic sites.
These gastrointestinal distribution profiles of free and total PDs were well consistent with the plasma concentration profiles. Namely, as to MP/EuL, PD was not distributed in the gastric and upper intestine long, leading to
 (a)
(a)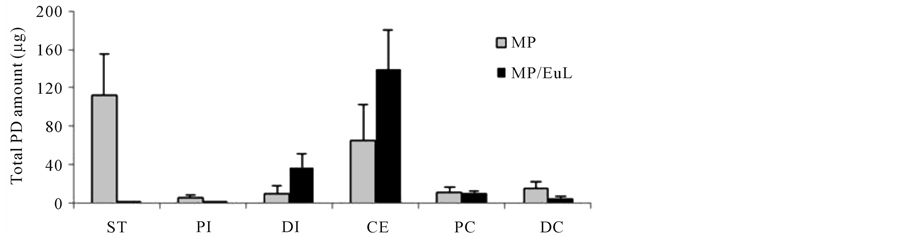 (b)
(b)
Figure 5. Gastrointestinal distribution of free (a) and total (free + conjugated) PD (b) 8 h after intra-gastric administration to rats with TNBS-induced colitis. The data of MP/EuL were based on the previous report [25] . The results are expressed as the mean ± S.E. (n = 3).
the low plasma concentration and short residence time in the systemic circulation. In addition, MP/EuL was located mostly in the area of DI, CE and colon, indicating that PD could be released only near the diseased site which was little involved in drug absorption.
On the other hand, after the dosing of MP, free PD still remained in the ST to a fair extent 8 h after the administration and to a certain extent in the PI area (Figure 5(a)), resulting in prolonged absorption of PD. As MP remained in the ST area to a fairly high extent (Figure 5(b)), PD was considered to be released from the ST area, though the release rate was very slow. These distribution characteristics of MP were considered to be due to the cationic charge of chitosan molecules.
As shown above, although MP and MP/EuL could deliver PD to the diseased site for a prolonged period, MP allowed the drug to be absorbed very slowly for a long time, while MP/EuL did not. In conclusion, MP/EuL were evaluated to be superior to MP for effective targeting to ulcerative colitis. It is concluded that entericcoating is very important for the targeting system using MP.