The Compatibility of Groups Used to Protect Phenolic Functionality during Oxone-Mediated Oxidative Esterification ()
1. Introduction
Chemoselective reactions occurring at the intended reactive sites are vital for the success of chemical transformations in a sequence. Protecting groups often play an essential role in organic synthesis, particularly for multi-step synthesis or for natural product total synthesis. Various protective agents are available to mask vulnerable functionality such as those for phenolic hydroxy groups. Indeed, a number of protective agents for phenolic hydroxy groups have been introduced over the past decades [1]. Among them, versatile groups include ethers, silyl ethers, esters, carbonates, and sulfonates. Previously, we reported a couple of chemoselective deprotection transformations from hydroxy moieties, in which trichloroethoxycarbonyl groups and trichloroacetyl groups were removed from aliphatic hydroxy groups and from phenolic hydroxy groups in the presence of indium powder [2]. Also, we reported a fast and practical approach to tetrahydropyranylation and depyranylation of alcohols using indium (III) triflate as the catalyst [3], which consequently was followed by a one-step transformation of tetrahydropyranyl ethers using catalytic indium (III) triflate [4]. Meanwhile, we recently demonstrated a practical method for the oxidative esterification of aldehydes using Oxone® monopersulfate compound (Oxone) as an oxidant with a catalytic amount of indium (III) triflate. Oxone is a versatile triple salt of potassium composed of potassium peroxymonosulfate. As for the starting materials, benzaldehyde derivatives were examined initially [5], and then application was expanded to heterocyclic aldehydes such as pyridinecarboxaldehydes [6]. In many cases, not only methanol but also longer chain alcohols could efficiently function as both the solvent and the substrate [7]. However, we soon realized that atert-butyldimethylsilyl (TBDMS) group [8] [9] that was intended to protect the phenolic hydroxy unit was removed during the reaction course of Oxone-mediated oxidative esterification. The para-toluenesulfonyl (Tosyl (Ts)) group [10], however, was maintained under the reaction conditions [5]. Based on these observations, we further investigated the compatibility of the protected phenolic functionality upon implementation of indium (III) triflate-catalyzed oxidative esterification using Oxone in methanol. A wide range of protecting moieties was selected and subjected to Oxone-mediated oxidative esterification. The details of this study are provided.
2. Results and Discussion
2.1. Preliminary Investigation
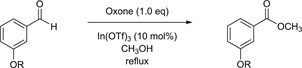
In our previous study, the TBDMS group that was intended to protect the phenolic hydroxy unit of meta-hydroxybenzaldehyde was removed by Oxone-mediated oxidative esterification [5]. Therefore, our first goal was to determine if silyl protection is always susceptible to reaction conditions such as these. While ortho and para substitution could feasibly display both inductive and resonance effects, meta substitution mainly shows only the inductive version. Thus, in order to eliminate the possibility of untoward resonance effects, derivatives containing a bulkytert-butyldiphenylsilyl (TBDPS) group for the protection of meta-hydroxybenzaldehyde, in addition to a TBDMS group, were envisioned and subjected to this type of Oxone-mediated oxidative esterification (Table 1, entries 1 and 2). Although Oxone, as the form of white granules, did not dissolve in the solution completely, the reactions flawlessly proceeded to the end. Fortunately, the TBDPS group showed stability against the reaction conditions, and the corresponding methyl ester was obtained in an 89% yield with the TBDPS group remaining intact. The TBDMS group, however, was removed during this reaction, as we previously reported. Since the acidity of the reaction mixtures could be the reason for deprotection, either acidic Oxone as the oxidant or indium (III) triflate as the Lewis acid could have been directly responsible for the deprotection. We then planned a further investigation, and reactions were carried out without either Oxone or indium (III) triflate (Table 1, entries 3 and 4). Unexpectedly, both reactions without Oxone and without indium (III) triflate disconnected the protecting TBDMS group, leaving meta-hydroxybenzaldehyde in a 95% yield and meta-hydroxybenzoic acid methyl ester in a 79% yield, respectively. Apparently, Oxone and indium (III) triflate are equally responsible for removing the protection for phenolic hydroxy groups.
2.2. Stable Protecting Groups under the Reaction Conditions
We then shifted our attention to scrutinizing a wide range of protecting groups. Various protective derivatives were prepared by known methods. Table 2 shows the results of the Oxone-mediated oxidative esterification of protecting groups that were sufficiently stable to resist cleavage under the reaction conditions. A triisopropylsilyl (TIPS) group, another bulky silyl group, was stable and furnished the desired methyl ester in an 81% yield (Table 2, entry 1). When phenolic hydroxy groups were protected by benzoyl (Bz) and benzyl (Bn) groups [11] [12], the reaction also proceeded smoothly and gave corresponding methyl esters in high yields (Table 2, entries 2 and 3) [13]. Similar to our previous observation of the inertness of a Tosyl (Ts) group under these reaction conditions [5], we sought to confirm the constant stability of sulfonates by further examining 2-nitorobenzenesulfonyl (Ns) and benzylsulfonyl groups. All experiments started with sulfonates and proceeded smoothly giving methyl esters with the sulfonate units intact, although the yields varied from 69% to 86% (Table 2, entries 4-6). Moreover, the Oxone-mediated oxidative esterification reaction did not disturb the 2,2,2-trichloroethoxycarbonyl (Troc)-protected carbonate [2] and gave methyl ester in a reasonable 78% yield (Table 2, entry 8).

![]()
Table 1. Reactions starting with silyl ethers as the starting materials.
a: All reactions were carried out at reflux in CH3OH. b: Isolated yields.
![]()
Table 2. Esterification reactions using the starting materials with stable protecting groups.
a: All reactions were carried out at reflux in CH3OH. b: Isolated yields.
![]()
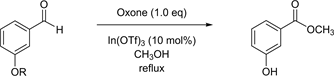
Table 3. Esterification reactions using starting materials with unstable protecting groups.
a: All reactions were carried out at reflux in CH3OH. b: Isolated yields.
2.3. Unstable Protecting Groups under the Reaction Conditions
Table 3 lists the entries from the Oxone-mediated oxidative esterification that were revealed to have started with compounds comprised of unstable protecting groups. In all cases during esterification, the protective moieties were cleaved, which furnished 3-hydroxybenzoic acid methyl ester. The tert-butoxycarbonyl (Boc) group is one of the most popular protecting groups and is known to be unstable particularly under acidic conditions [14]. Indeed, although methyl esterification was implemented smoothly, deprotection simultaneously proceeded, and the reaction gave the deprotected methyl ester in a 98% yield (Table 3, entry 1). We examined other protecting groups with ambiguous stability against acidic environments, such as tetrahydropyranyl (THP) [3] and acetyl (Ac) groups. In both cases, the reactions did not sustain the protective agents, and 3-hydroxybenzoic acid methyl ester was generated in high yields (Table 3, entries 2, 3). Consequently, it occurred to us that the bulkiness of acid-sensitive acetals might improve the stability of the protective elements. Thus, we added heavy and long methoxyethoxymethyl (MEM) and benzyloxymethyl (BOM) protective agents [15], as well as smaller ones such as ethoxymethyl (EOM) and methoxymethyl (MOM) agents [16] [17] [18]. Despite our blurred predictions, the resultant products that started with these four acetals were identical, and 3 hours of reaction time at reflux produced a deprotected methyl ester (Table 3, entries 4-7). A minor difference was noted when the heavy and long acetals gave higher yields (Table 3, entries 4, 5) than the smaller ones (Table 3, entries 6, 7).
3. Conclusion
We investigated various groups that could provide protection for phenolic hydroxy groups when Oxone-mediated oxidative esterification was implemented. The reactions employed Oxone and a catalytic amount of indium (III) triflate in methanol. Under these reaction conditions, stable protecting groups remained intact during esterification as a part of the starting materials and the products, whereas unstable protecting groups were cleaved during esterification. Sulfonates were stable and compatible during the reaction courses. On the other hand, acetals were unstable and incompatible. These findings should help guide the choice of proper protection for phenolic hydroxy groups.
4. Experimental
4.1. Materials and Instruments
All reagents were of analytical grade, were purchased commercially, and were used without further purification. All reactions were performed under argon using magnetic stirring unless otherwise stated. 1H NMR and 13C NMR spectral data were recorded on a JEOL JMTC-500 spectrometer (500 MHz for 1H NMR and 125 MHz for 13C NMR) using tetramethylsilane (TMS) as the internal standard.
4.2. General Experimental Procedure
When used as the starting materials, benzaldehyde derivatives such asmeta-tert-butyldiphenylsilyloxy benzaldehyde (360 mg, 1.0 mmol) were combined with Oxone (615 mg, 1.0 mmol) and indium (III) triflate (56 mg, 10 mol%) in methanol (50 mL). The reaction mixtures were heated at reflux and monitored for completion via TLC. The reaction mixtures were filtered, and the filtrate was condensed by rotary evaporation. The resultant residue was purified by silica gel flash column chromatography to obtain the desired methyl ester products, which were confirmed by spectroscopy.
3-tert-Butyldimethylsilyloxybenzoic acid methyl ester (Table 1, Entry 1): 1H NMR (500 MHz, Chloroform-d) δ 7.78 (dd, 4H,J = 8.1, 1.8 Hz), 7.61 - 7.59 (m, 2H), 7.46 (t, 2H,J = 7.5 Hz), 7.41 (t, 4H,J = 7.5 Hz), 7.12 (t, 1H,J = 8.1 Hz), 6.91 (dt, 1H,J = 8.1, 1.8 Hz), 3.87 (s, 3H), 1.18 (s, 9H); 13C NMR (125 MHz, Chloroform-d) δ 166.7, 155.5, 135.4, 132.4, 131.3, 130.0, 129.0, 127.8, 124.1, 122.2, 120.8, 51.9, 26.4, 19.4.
3-Triisopropylsilyloxybenzoic acid methyl ester (Table 2, Entry 1): 1H NMR (500 MHz, Chloroform-d) δ 7.61 (dt, 1H, J = 7.8, 1.2 Hz), 7.54 (dd, 1H,J = 2.6, 1.4 Hz), 7.27 (t, 1H,J = 8.0 Hz), 7.06 (ddd, 1H,J = 8.1, 2.6, 1.2 Hz), 3.89 (s, 3H) 1.27 (sept, 3H,J = 7.7 Hz), 1.11 (d, 18H,J = 7.7 Hz); 13C NMR (125 MHz, Chloroform-d) δ 166.9, 156.0, 131.3, 129.2, 124.5, 122.2, 120.7, 52.0, 17.8, 12.5.
3-Benzoyloxybenzoic acid methyl ester (Table 2, Entry 2): 1H NMR (500 MHz, Chloroform-d) δ 8.20 (dd, 2H, J = 8.3, 1.4 Hz), 7.96 (dt, 1H, J = 7.8, 1.4 Hz), 7.91 (t, 1H,J = 1.7 Hz), 7.62 (tt, 1H,J = 7.5, 1.4 Hz), 7.51 - 7.47 (m, 3H), 7.42 (ddd, 1H,J = 8.0, 2.3, 1.2 Hz), 3.90 (s,3H); 13C NMR (125 MHz, Chloroform-d) δ 165.9, 164.7, 150.7, 133.6, 131.5, 130.0, 129.3, 128.9, 128.5, 126.9, 126.3, 122.8, 52.1.
3-(para-toluenesulfonyloxy)benzoic acid methyl ester (Table 2, Entry 4): 1H NMR (500 MHz, Chloroform-d) δ 7.89 (d, 1H,J = 7.8 Hz), 7.67 (d, 2H,J = 8.1 Hz), 7.63 (s, 1H), 7.34 (t, 1H,J = 8.0 Hz), 7.29 (d, 2H,J = 8.0 Hz), 7.15 (dd, 1H,J = 8.0, 1.4 Hz), 3.85 (s, 3H), 2.41 (s, 3H); 13C NMR (125 MHz, Chloroform-d) δ 165.5, 149.4, 145.6, 131.9, 137.8, 129.8, 129.6, 128.3, 128.1, 126.7, 123.4, 52.3, 21.6.
3-(ortho-Nitrophenylsulfonyloxy)benzoic acid methyl ester (Table 2, Entry 5): 1H NMR (500 MHz, Chloroform-d) δ 7.97 (dt, 1H,J = 7.5, 1.7 Hz), 7.94 (d, 1H,J = 8.1 Hz), 7.86 - 7.82 (m, 3H), 7.72 - 7.67 (m, 1H), 7.43 (d, 1H,J = 8.1 Hz), 7.40 (ddd, 1H,J = 8.0, 2.3, 1.2 Hz), 3.89 (s, 3H); 13C NMR (125 MHz, Chloroform-d) δ 165.4, 148.9, 148.6, 135.7, 132.2, 132.1, 132.0, 130.0, 128.7, 127.9, 126.7, 125.0, 123.2, 52.5.
3-Benzylsulfonyloxybenzoic acid methyl ester (Table 2, Entry 6): 1H NMR (500 MHz, Chloroform-d) δ 7.94 (dt, 1H,J = 7.8, 1.2 Hz), 7.74 (t, 1H,J = 2.0 Hz), 7.47 - 7.45 (m, 2H), 7.42 - 7.39 (m, 4H), 7.29 (ddd, 1H,J = 8.3, 2.3, 1.2 Hz), 4.55 (s, 2H), 3.89 (s, 3H); 13C NMR (125 MHz, Chloroform-d) δ 165.5, 148.9, 131.9, 130.7, 129.8, 129.2, 128.9, 128.1, 126.8, 126.5, 122.9, 56.9, 52.3.
3-(2’,2’,2’-Trichloroethoxycarbonyloxy)benzoic acid methyl ester (Table 2, Entry 7): 1H NMR (500 MHz, Chloroform-d) δ 7.96 (dt, 1H, J = 8.0, 1.7 Hz), 7.89 (t, 1H,J = 1.7 Hz), 7.48 (t, 1H,J = 8.0 Hz), 7.41 (ddd, 1H,J = 8.0, 2.3, 1.2 Hz), 4.87 (s, 2H), 3.91 (s, 3H); 13C NMR (125 MHz, Chloroform-d) δ 165.6, 152.1, 150.5, 131.7, 129.5, 127.5, 125.2, 121.9, 93.8, 76.6, 52.2.