1. Introduction
Petroleum is known to be the mean source of fuel energy all over the world and will remain so until at least a few decades. As a means of reducing environmental contamination to coincide with the widely adopted legislations for sulfur and nitrogen, it is important to remove sulfur compounds from middle distillate fuels, typically using hydrodesulfurization (HDS). However, polyaromatic sulfur species constitute the major portion of the sulfur containing compounds in this fuel and are difficult to fully degrade via this catalytic process [1] - [6]. During HDS, the activity of catalysts is affected by the presence of various reaction matrixes, and H2S is one that is known to greatly slow down the rate of the reaction by inhibiting the hydrogenolysis of C-S bonds [1] [2]. Nevertheless, under such conditions, the sulfur substrate species must be reacted under the presence of H2S. Many studies based on either fixed bed systems or batch reactors have reported drastic suppression of the catalysis of the HDS reaction involving both model sulfur compounds and real feed stocks in the presence of H2S [7] [8] [9] [10]. Such behavior is well-known in this area of research where the consensus catalysts are subjected to seriously lose their overall activity because of H2S. The extent of activity reduction is varied and highly dependent on the catalyst identity where the established reaction network of sulfur substrate is important. However, interesting results concerning the HDS of dibenzothiophene (DBT) and 4,6-dimethyldibenzothiophene were recently reported [11] [12], which showed that H2S may actually promote the HDS activity of some certain MoS2 class catalysts. To the best of our knowledge, such results are interesting and yet there is surprisingly a lack of literature on following and widely investigating this point. Thus, kinetic modeling that thoroughly describes this approach is needed. The overall catalytic activity and selectivity appear to be dependent on the concentration of H2S in the reaction media. Overall, it is evident from the results to date that H2S somehow modifies the chemical nature of the active sites in this reaction. To investigate the catalyst activity promotion phenomenon by H2S and to determine the adsorption equilibrium constants, the present work performed a number of DBT HDS experiments in conjunction with a MoS2 catalyst under various conditions, and derived detailed kinetic models to assist in understanding the reaction mechanism.
2. Experimental
A model MoS2 catalyst was prepared by a heat treatment procedure previously described in the literature [11]. Briefly, a precursor of ammonium heptamolybdate tetrahydrate (NH4)6Mo7O24·4H2O were subjected to a gradual thermal annealing till reaching to 850˚C under a stream of H2S/H2 (10% v/v) gas. After synthesis, the catalyst was milled using zirconia balls and then sulfided at 400˚C and 1 atm under a 10% v/v H2S/H2 gas mixture flowing at 100 sccm in preparation for the experimental trials. All reactions were carried out in a 100 ml batch-type micro-autoclave from which aliquots were withdrawn at specific time intervals, using 100 mg of the catalyst for each trial. The feed consisted of 1 wt% DBT in decane although, in some experiments, 500 mg Cu powder was also added as a H2S scavenger to establish the initial kinetics without matrix interference [13]. The use of this type of reactor allowed the effects of the reaction matrix and operating conditions to be examined. All reactions were performed at 340˚C and an initial H2 pressure of 3 MPa. The H2S concentration in the reaction mixture was changed by adding various quantities of S powder to the autoclave. This S was converted quantitatively to H2S by reaction with H2, as confirmed by a blank test according to the following equation.
3. Results and Discussion
The major products detected following the HDS of DBT were biphenyl (BP), cyclohexylbenzene (CB), partially hydrogenated DBT (HDBT) and H2S. As noted, various H2S concentrations were employed during the HDS reaction, and Figure 1 and Figure 2 summarize the intermediate products obtained from hydrogenation (HYD) and direct desulfurization (DDS) reactions during the HDS of DBT. The formation of these products depended on whether the H2S that was generated in situ was removed from or retained in the reaction mixture. Specifically, HYD products were generated in greater proportion in the presence of H2S, while the DDS products were decreased. It is interesting to note from Figure 2 the non-linear nature of the HYD route products.
![]()
Figure 1. Product distributions during the HDS of DBT in the absence of H2S.
![]()
Figure 2. Product distributions during the HDS of DBT in the presence of H2S.
3.1. Kinetics
Several assumptions and simplifications were involved in the kinetics calculations. Firstly, the HDS of DBT was assumed to proceed primarily by DDS and HYD, as in Scheme 1.
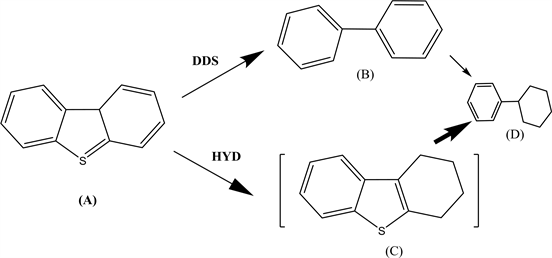
Scheme 1. Intermediate products formed during the HDS reaction of DBT.
In addition, CB was assumed to result mainly from partially-hydrogenated DBT, and so the formation of this product will be promoted by H2S, in agreement with prior reports [5] [6]. The effect of the H2 pressure on the reaction rate was neglected because H2 was added in excess and so could be included in the intrinsic rate constant. Two active sites were considered, one for HYD and the other for DDS, according to the equations (all symbols and parameters are identified either in the notation section or in the text):
, (1)
(2)
and
. (3)
In this case, the concentration of hydrogenation sites (πH) will be affected by the presence of H2S while the concentration of σD sites remains almost unaffected. Two scenarios were considered, consisting of the HDS reaction of DBT with and without H2S.
3.2. Kinetic Model for HDS with a Negligible H2S Concentration
Some prior studies have shown that the HDS of DBT is a pseudo-zero-order reaction [14] [15], while others have determined that this is a pseudo-first-order reaction [7] [12] [16] [17] [18]. As discussed below, the results of the present study agree with the latter behavior and so the analysis in this paper is predicated on pseudo-first-order kinetics. Based on mass balances of the reaction components, rate equations were adapted to quantitatively describe the product distributions. These equations (details of which have been previously published [19]) are:
(4)
(5)
and
(6)
The reaction rate in the absence of H2S was well represented by the pseudo-first order relationship in Equation (4), and a plot of the data is presented in Figure 3. Here the fit of the experimental points to this equation is confirmed by the high regression factor. Equations (5) and (6) were used to fit the experimental results for the reaction intermediates, which required the iteration estimation of only two variables (k1 and k2). Each data fitting was performed using the Mathcad software package and employing non-linear methods via sequential iterations until convergence. Figure 4 plots the concentrations of the intermediate species against reaction time and also shows the fits obtained using Equations (5) and (6), which confirm the validity of the model. Fitting the experimental data with the model allowed the rate constants to be determined, which in turn enabled an estimation of the contribution of intermediates B and C to
![]()
Figure 3. Pseudo-first order plots of data acquired during the DBT of HDS in the absence of H2S at 340˚C and 3 MPa H2.
![]()
Figure 4. Experimental data and fittings for the generation of BP and H4-DBT during the HDS of DBT in the absence of H2S.
the yield of D during the HDS reaction. The fractions of B or C transformed to D at a given time t were estimated from the equations:
(7)
and
(8)
The apparent kinetic parameters determined in this manner are provided in Table 1. The model used in this work was based on a version of the Langmuir-Hinshelwood mechanism, and so the rate equations for the HYD and DDS reactions were:
(9)
and
. (10)
By substitution, it is possible to obtain the equations:
(11)
(12)
and
. (13)
Once the individual apparent rate constants were found, the equilibrium adsorption constants could be compared. The experimental data were fit according to Equations (11)-(13), as shown in Figure 5. Using the apparent rate constants for the parallel reactions determined from the previous calculations, the adsorption equilibrium constants for the substrate on the σ and π sites were predicted and the values are provided in Table 2.
3.3. Kinetic Model for HDS in the Presence of H2S, Promotion Model
Data regarding the product distributions are the primary means of validating
![]()
Table 1. Kinetic parameters for the HDS of DBT at various H2S concentrations.
*Individual apparent rate constants, with an uncertainty of approximately 3% - 5%.
any kinetic model, and typical reversible behavior was observed in the present case both with and without H2S. This was ascertained by experimental trials during which the reaction was performed using varying concentrations of H2S. Under these conditions, the promotional effect of the H2S was determined to involve a physical reversible process and was associated with the nature of the catalyst. In the existing of H2S, the hydrogenation reaction products were predominant, as can be seen by comparing Figure 4 and Figure 6, and so the activity of this route was obviously increased. The effect of H2S can be discussed in terms of its concentration in the reaction feedstock. Because the reaction is carried out in a batch system, a classical treatment of the autocatalytic-like kinetics leads to the equations:
(14)
and
(15)
![]()
Figure 5. Experimental data acquired during the HDS of DBT in the absence of H2S and fittings based on the Langmuir-Hinshelwood relationship in Equation (13).
![]()
Table 2. Equilibrium adsorption constants and kinetic coefficients determined from model fittings for data acquired during the HDS of DBT.
a: s−1∙gcat−1.
![]()
Figure 6. Experimental data for the generation of BP and H4-DBT during the HDS of DBT in the presence of 17 × 10−4 moles H2S.
Here, Rt is the total reaction rate and kts is the overall rate constant under this conditions. In the present trials, the sum total of the moles of DBT and H2S at any given time remained constant, and so we can write:
(16)
Equation (15) can be rearranged after integration to:
(17)
Figure 7 depicts the experimental data according to this equation, and demonstrates suitable linear regression. Based on the Langmuir-Hinshelwood mechanism, if the H2S that is generated in situ is retained in the reaction medium, we can write the rate (RH-S) as follows:
(18)
which, after rearrangement and substitution, becomes:
(19)
The DDS route is not considered to be affected by the presence of H2S, and so we have:
(20)
This indicates a special case in which the reaction rate exhibits autocatalytic-like behavior. In an autocatalytic process, one of the reaction products is involved in the catalytic mechanism and so should be included in the rate law. During HDS over the present MoS2 catalyst, the reaction rate was found to be promoted by H2S, and thus this species may affect both the potential and the nature of the active catalytic sites. Therefore, the overall rate in the presence of H2S that is generated in situ, Rts, can be represented as:
![]()
Figure 7. A linear plot confirming the autocatalytic nature of the HDS of DBT, based on Equation (17).
(21)
where Rts is the overall reaction rate. The correlation associated with Equation (21) is depicted in Figure 8 and shows a suitable fit to the experimental data. Based on the constants estimated from both the classical treatment and the data analysis using Equations (19)-(21), the adsorption equilibrium constant associated with H2S can be determined, and the results of such calculations are provided in Table 2.
3.4. Analysis of the Kinetic Data
In model simulations, as the constants utilized in the model remain limited in number then the validity of model to be adopted increased. To minimize the number of constants included in our kinetic model, we first calculated reaction parameters based on the proposed model of Equations (5) and (6), and then estimated the fraction of each intermediate product that participates in the reaction, followed by manipulating the kinetic treatments once more according to the promotion model. From these calculations, the ratio of the DBT adsorption equilibrium constant for π sites to that for σ sites was found to be close to unity. Thus, there was no preferential adsorption of the reactant on either active site when using the present catalyst. It should be noted that the adsorption equilibrium constants were estimated to have this limited value Kσ or Kπ ≤ 0.01 and that the ratio of Kπ/Kσ ≈ 1. Based on prior research regarding HDS reactions and the kinetic parameters estimated in this study, it is possible to analyze the experimental data as follows.
3.4.1. First Stage
When the HDS reaction was performed in the absence of H2S, the data could be fitted with a model recently developed by our group in which all the reaction products are included in the rate equations (that is, Equations (4)-(6)). This model was subsequently used to simulate the experimental data shown in Figure 4 and a reasonable agreement between the experimental values and the simulation can be seen. The estimated values predicted by this model are presented in Table 1. If the HDS process proceeds as a pseudo-first-order reaction, then this
![]()
Figure 8. Experimental data acquired during the HDS of DBT in the presence of H2S with fitting based on Equation (21).
model may be applicable.
3.4.2. Second Stage
If the H2S generated in situ remains in the reaction mixture, a kinetic rate curve corresponding to autocatalytic behavior is obtained, as is evident in Figure 7 and Figure 8. In this case, models based on Equations (17) and (21) were applied to simulate the experimental data for the overall reaction, and these predictions matched the trend exhibited by the experimental values. The equilibrium constants for substrate adsorption on the two active sites and their ratios should certainly be considered primarily on applying this model. In addition, the rate constants for the two parallel routes (i.e., DDS and HYD) should be taken into consideration when fitting data acquired in the presence of H2S. For this reason, the estimated HDS kinetic parameters for the reaction in case of absence of H2S were utilized to assess the reaction in the presence of H2S. Including such limits may help improve the simulation results. The results clearly showed that there was little change in the DBT and H2S adsorption equilibrium constants and that the values of these constants remained ≤0.01.
3.4.3. Third Stage
In presence of an excess of H2S, a part of this species modifies the potential active sites while the rest is essentially not involved in the reaction. This scenario is quite similar to that in an actual refinery performing HDS. The catalyst may be involved in the reaction in a manner which is kinetically similar in nature to that found in the first stage section. The model discussed above in relation to the first stage can also be applied to describe this reaction, and the associated kinetic data are listed in Table 1. It is interesting to observe that the H2S has an effect following the first dose, such that the reaction proceeds as though the H2S undergoes self-promotion. Thereafter, with increases in the H2S concentration in the feedstock, its primary effect remains almost unchanged and the reaction behaves kinetically as a pseudo-first-order process. This effect can possibly be attributed to the formation of new active sites created by the H2S. After an equilibrium is achieved, at which point the H2S has modified all possible potential active sites, the reaction proceeds normally through the participation of the two kinds of active sites. Thus, the self-promotion effect induced by H2S is basically temporary and is associated with the modification of active sites up to saturation. Another point to note is that an excess of H2S induces neither further promotion nor inhibition of the catalytic activity, Figure 9. These results emphasize that H2S acts as a promoter possibly by increasing the efficiency of active sites, by increasing the number of sites, or by both mechanisms.
3.5. Inhibition and Promotion
At present, there is a lack of full information regarding the effect of compounds such as H2S and organic nitrogen compounds on catalytic behavior during HDS over unsupported bulk MoS2 catalysts. The presence of H2S evidently inhibits the
![]()
Figure 9. Catalytic activity versus the H2S amount in the feedstock.
DDS reaction of polyaromatic refractory sulfur containing compounds but has little impact on the HYD pathway. This detrimental effect on activity is apparent when employing conventional catalysts. As an example, an inhibition effect was reported in reactions using conventional CoMo/Al2O3 catalysts, and was also reported to be reversible [7]. The present results concerning the MoS2 catalyst show a remarkable enhancement of the HYD route upon the addition of H2S. Two kinetic models are proposed: one in the absence of H2S and the other with H2S in the reaction medium at moderate levels. H2 appears to be essential to the HDS process and may play a specific role in modifying the potential active sites that become two independent types: for the HYD and DDS reactions. The experimental results in the absence of H2S can be reasonably described by the model based on Equations (4)-(6). In contrast, in the presence of H2S, the occurrence of HYD is promoted and the overall catalytic activity is enhanced. This improvement in the activity is obviously due to the increased rate of the HYD reaction. The effect of H2S on kinetics leads to the hypothesis that H2S interacts chemically with the catalyst by creating new highly active HYD sites. The good agreement of the experimental results with the non-linear numerical fitting supports this hypothesis.
Thus, H2S is believed to act as a reagent to create a fixed number of potential HYD active sites. These sites can be modified either by H2 or H2S. While H2 modification of the active sites leads to two distinct types of sites for the two routes, H2S modifies the active sites to selectively promote the HYD reaction. The efficiency of the HYD active sites created by H2S is likely much more pronounced than those generated by H2. The present results and the fitting models also suggest the potential for mutual interconversion of the newly created active sites. Egorova et al. [5] reported that H2S has a positive effect on the cleavage of C-N bonds. It is unclear if this effect is due to the sulfidation state of the catalyst or to the participation of H2S in the reaction mechanism, although the authors suggest the latter. The present study supports this hypothesis too. This unique behavior of H2S in conjunction with this particular bulk MoS2 catalyst during HDS is opposite to the behavior reported with conventional catalysts. These results confirm the importance of the structure of the MoS2 species, and suggest that not all such species will show similar effects together with H2S.
4. Conclusion
Kinetic models based on the assumption of two distinct active sites for DDS and HYD during the HDS of DBT over a MoS2 catalyst were constructed and exhibited good agreement with experimental results. According to these models, H2S promotes HYD via its chemical involvement in the reaction mechanism, while H2 produces two distinct active sites: one for HYD and the other for DDS. The presence of H2S might lead to the formation of new and highly effective HYD active sites. Because of the fixed number of active sites that can be activated either by H2 or H2S, it is important to consider the quantity of newly created sites as a ratio of the total number of sites when assessing selectivity. Thus, as HYD active sites (π*) are increased in number, the quantity of DDS active sites (σ) decreases. Overall, this study suggests that two reaction mechanisms are possible, depending on whether or not H2S is present in the reaction medium. It can be inferred that the HDS reaction can be promoted by including an optimal H2S concentration in the reaction feedstock so as to create more active sites.
Acknowledgements
This work was supported by Kyushu University, Japan, and Mansoura University, Egypt.
Notation
Active hydrogenation sites generated by H2S
Active hydrogenation sites generated by H2
Active direct desulfurization sites
Potential active sites
k1 Rate constant for the hydrogenation of DBT
k2 Rate constant for DDS
k3 Rate constant for BP hydrogenation
k4 Rate constant for the hydrogenation of DBT
k˚ Sum of k1 and k2
Kπ Equilibrium constant for the adsorption of DBT on π sites
Kσ Equilibrium constant for the adsorption of DBT on sigma sites
KπS Equilibrium constant for the adsorption of H2S on π sites
kπ Intrinsic rate constant for hydrogenation
kσ Intrinsic rate constant for DDS
kπs Intrinsic rate constant for hydrogenation in the presence of H2S
kt Overall rate constant in the absence of H2S
kts Overall rate constant in the presence of H2S
CD Concentration of DBT at time t
CS Concentration of H2S
CH Concentration of HDBT
CB Concentration of BP
CD˚ Initial concentration of DBT
CS˚ Initial concentration of H2S
RH-S Rate based on Langmuir-Hinshelwood mechanism