Docking of Glycokinase with Oxo, Sulfo, and Seleno Derivatives of the Carboxamide Activator S41 ()
1. Introduction
Glucokinase (GK) belongs as hexokinase to essential proteins which play a significant role in the animal world [1]. Indeed, glucokinase is one of four hexokinases present in high proportion in some organs such as the liver and pancreas, where it is involved in the hepatic metabolism of glucose and the pancreatic secretion of insulin [2] [3] [4]. The high level of glucose concentration in blood and urines associated with diabetes caused in part by inactivation of the gene encoding glucokinase [2].
Due to weak selectivity and side effects of the commonly used antidiabetic drugs, there is a growing interest in new pharmacological strategies requiring less time and capital to improve the antidiabetic therapy [2]. Disadvantages of current strategies based on experimental screening of molecules to identify any that elicit desired biological response are low rate and assays needing extensive development with validation before use. One of the new approaches is the computer-aided drug discovery (CADD) based on molecular docking in conjunction with molecular dynamic simulation and quantitative structure activity relationship (QSAR). Ligand-protein docking has been used to probe pharmacophoric potential of ligands interacting at the activator site of glucokinase [5] - [14]. A comparative investigation on activator potential of numerous pharmacophores revealed that amide ligands consisting of three flexible cyclic arms joined in Y letter shape showed the highest binding affinity at the GK allosteric site [6].
In the present work, we used the derivatives of N,N-dimethyl-5-(2-methyl-6-((5-methylpyrazin-2-yl)-carbamoyl)benzofuran-4-yloxy)pyrimidine-2-carboxamide (S41), which show a shape resembling to the Y letter, in order to explore their ability to interact at the allosteric site of GK using molecular docking. The docking was carried out with the GOLD 5.6 program (Genetic Optimization for Ligand Docking) implemented in the Cambridge Structural Database (CSD). Gold fitness scores as well interaction energies were calculated to determine the affinity and the thermodynamic stability of the binding, respectively [15] [16]. The binding affinity of the S41 activator was compared with those of oxo, sulfo and seleno derivatives. GK was derived from the Protein Data Bank (PDB code: 3S41). Das et al. [17] showed that the affinity of a ligand to the GK activator site was modified by replacing an atom or a group of atoms inducing marked changes in ligand hydrophobicity and hydrophilicity. Kilembe et al. (2019) also reported on changes of ligand affinity to N-terminal domain of Heat shock protein 90 (HSP90) when oxygen atoms were replaced by sulfur or selenium atoms [18].
2. Materials and Methods
2.1. Preparation of 3S41
The crystal structures of the glucokinase complex with the co-crystallized ligands used in the present study were retrieved from the Protein Data Bank (PDB code 3S41) and imported into HERMES 1.6 visualization interface implemented in the Cambridge Structural Database (CSD). Survey of the activator binding site was performed with the reference of amino acid residues of large GK domain as previously reported [9] [14] . Hydrogen atoms were then added to the protein for correct ionization and tautomeric states of amino acid residues. The molecular docking of 3S41 was carried out with GOLD 5.6 after conversion of its structure from 2D to 3D without minimization of protein energy.
2.2. Preparation of Ligands
The carboxamide ligand was the S41 co-crystallized activator of GK. One fragment of S41 structure was modified as labelled in Scheme 1 in order to design its 30 seleno, sulfo and oxoderivatives using Mercury 3.10 program implemented in CSD.
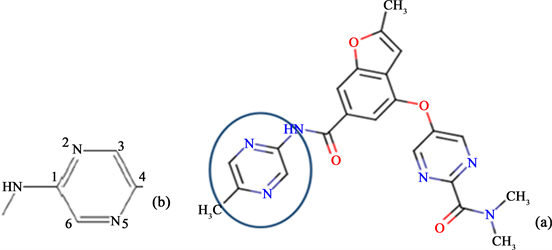
Scheme 1. (a) Structure of the S 41 co-crystallized ligand and fragment of the structure modified to obtain seleno, sulfo and oxo derivatives of S41 (b).
Table 1 reports modified structure fragments and ligand reference codes.
![]()
![]()
Table 1. Modified fragments with reference codes of the ligand structures.
Mercury 3.10 program was used to generate and optimize ligand conformations after the conversion of their structures from 2D to 3D. Energetically least conformers were selected for further use. The calculation of ligand molecular electrostatic potentials were performed at the Hartree-Fock theory (HF), implemented in the Gaussian 09 program [19], using the 6 - 31 G basis set.
2.3. Molecular Ligand-Protein Docking
The ligand-protein docking was carried out using the GOLD 5.6 program. Before docking GK with ligands, the reproduction of the experimental poses of S41 using 3S41 and 24 additional GK complexes with their co-crystallized ligands derived from PDB as shown in Table 2 was carried out to validate GOLD 5.6. A good reproduction was defined as one with a value of the root mean square deviation (RMSD) lower or equal to 2 Å [20] [21].
![]()
Table 2. PDB codes and resolutions (R) of the GK complexes used for validation of GOLD 5.6.
The default GOLD fitness function ChemPLP was used for the docking of GK-3S41 with derivatives by considering the flexibility of the ligands and specific amino acid residues in the activator site. The greater the ChemPLP fitness score, the better is the ligand binding affinity. Rescoring with Chemscore was performed to determine the energy of the ligand interaction with the GK site (ΔGbind) according Scheme 2.
Scheme 2. Free enthalpy of binding, where the ΔG0 and S (bar) components are experimental and entropic terms, while ΔGHB, ΔGMet, ΔGLip, ΔGRot represent energy terms related to hydrogen-bonding, metal, lipophilic and rotation interactions, respectively [22].
Each simulation was performed ten times providing ten docked conformations until three of the ten poses were within 1.5 Å RMSD of each other. The ligand conformer pose with the lowest energy was considered as the binding conformation in the protein site.
3. Results and Discussion
3.1. Validation of GOLD 5.6 Potential for Ligand-Protein Docking
Figure 1 and Table 3, respectively, report the pose of the S41 in the activator site derived from PDB and the pose provided by GK docking with the GOLD 5.6 program.
![]()
Figure 1. (a) 3D structure of the 3S41 complex [14]; (b) Pose of the co-crystallized S41 reproduced by GK docking with GOLD 5.6.
![]()
Table 3. Experimental pose of the S41 ligand derived from PDB and its pose with geometric patterns of the specific interactions provided by docking using GOLD 5.6.
Figure 1 as well as Table 3 show that the experimental pose of the co-crystallized ligand (S41) derived from PDB is well reproduced by docking with the GOLD 5.6 program. Indeed, the pattern of the ligand pose in Figure 1 matches well the activator site with its three hydrophobic pockets in form of the Y letter [6]. The docking reproduces well the hydrogen bonding interactions with their respective distances (δ) and angles (θ). The same (Ar63))O---HN bond distance of 2.09 Å was found for both 3S41 derived from PDB and 3S41 from modelling with GOLD 5.6. A weak deviation of 0.24 Å (12%) was observed for theArg63)NH---N bond. Smaller deviations of 3˚ (2%) and 7˚ (4%) were found for angle values related to the first bond and the second one, respectively.
The distribution of RMSD values derived from the pose reproductions of co-crystalized activators using 25 examples of GK complexes (Figure 2) also supports the high potential of GOLD 5.6 for ligand-protein docking.
![]()
Figure 2. Distribution of RMSD values derived from the reproduction of experimental poses using 25 complexes of GK with co-crystallized ligands. 88% of the poses (in blue) were reproduced with RMD ≤ 2 Å; 12% (in red), with RMD > 2 Å.
Figure 2 reveals that 22 experimental poses (88%) are reproduced with RMSD values ≤ 2 Å by the docking, while 3 experimental poses (12%) are reproduced with RMSD values > 2 Å. Thus, most of RMSD values are equal or less than the limit value recommended for a good pose reproduction [20] [21] suggesting that GOLD 5.6 is a powerful docking program [18] [22].
The experimental pose of co-crystallized activator from PDB (S41) and superposition of the poses derived from the molecular docking with GOLD 5.6 are illustrated in Figure 3.
![]()
Figure 3. Superposition of S41 poses in the binding site of GK: (a) An experimental pose (in magenta) and a pose derived from docking (in green) with GOLD 5.6 (RMSD = 0.32Å); (b) Typical superposition of S41 poses.
The RMSD of 0.32Å was obtained by superposing the ligand poses confirming the docking potential of the GOLD 5.6 program [18] [22].
3.2. Glucokinase Docking with Ligands
The areas of contacts between GK and ligands are illustrated in Figure 4.
![]()
Figure 4. GK complexes with contact areas involved in the binding of 3S (a) and (b) 3Se.
Figure 4 reveals that hydrogen bonds and hydrophobic interactions were involved in the ligand binding as expected. This finding is consistent as the activator site is a combination of three hydrophobic pockets [6], yet it contains residues of polar amino acids, which are able to form hydrogen bonds with ligands [23]. Indeed, the first pocket contains Val62, Ile59, Val452 and Val255, which are hydrophobic, and the polar Arg63. The second pocket contains Pro66, Val65 and Tyr215, while the third pocket contains Met210, Met235 and Tyr214. Ligand areas allowing hydrogen-bonding and lipophilic interactions were evidenced by the molecular electrostatic potential surfaces (MEPs) depicted in Figure 5.
![]()
Figure 5. Computed electrostatic potential on 0.001 a.u molecular surfaces of the ligands 3S (a), S41 (b) and 1Se (c).
It can be seen that MEPs of S41 and derivatives feature marked positive (in blue) and negative (in red) areas corresponding to hydrogen-bond donors or acceptors, but also large non-polar regions (in grey). Thus, lipophilic interactions are probably due to the contacts between hydrophobic amino acid residues present in the activator site of GK and non-polar areas of ligand molecules, while the hydrogen bonds can be ascribed to electrostatic interactions between hydrogen-bond donors and acceptors of polar amino acid (AA) residues and those of S41 and its derivatives. The affinity of the ligands to the binding site was then determined based on GOLD fitness scores, while the binding stability was obtained by calculating the energy of interaction [18]. Table 4 reports the values of GOLD fitness scores and energies of ligand-GK interaction.
![]()
Table 4. Gold scores, free enthalpy of binding and its components terms in kcal.mol-1 assuming that ΔG0 = -5.48 kcal.mol-1 and S =0 kcal.mol-1 [18] [22].
The values of GOLD fitness scores and free enthalpies of ligand binding suggest that the S41 activator and its derivatives were bonded with high affinity to GK. According to the fitness scores, 12 derivatives interact with GK more strongly than S41. All values of the binding enthalpies are negative suggesting that all ligand-GK complexes are thermodynamically stable. The binding enthalpies of the most stable complexes decrease in the sequence 3S> 5S > 1S >1O > 3O > 4O > 8S > 3Se. Values of energy component terms (ΔGHB and ΔGLip) show that the main forces stabilizing ligand-protein complexes are lipophilic interactions, enhanced by hydrogen bonding interactions. This is evidenced by the increase of ΔGLip when an oxygen atom in the ligand is replaced by a chalcogen atom (S or Se). Such hydrogen-binding and lipophilic interactions should improve the activator selectivity and lead to least side effects, when they involve specific amino acid residues [6].
The predominance of the lipophilic interactions can be ascribed to hydrophobic amino acid residues present in the activator site. Indeed, it has been found that the Glu221, Met235, Ile211, Val455, Tyr215, Val91, Ala454, Leu451, Tyr214 and Val 62 amino acid residues were involved in the lipophilic interactions. Val62 and Tyr 215, respectively, interacted with ligands in the first and second pockets of the activator site, while Met235 and Tyr 214 were involved in hydrophobic effects in the third pocket as shown in Figure 4. This finding is in good agreement with the literature [6]. Table 5 reports distances and angles related to hydrogen bonds found in the binding site for the 6 best ranked ligands. The geometric parameters were calculated according to Scheme 3. The value vdW (H) = 1.10 Å determined by Rowland et al. (1996) [24] was used for van der Waals radius of hydrogen atom, while the values vdW(O) = 1.52 Å for oxygen, vdW(N) = 1.55 Å for nitrogen and vdW(Se) = 1.90 Å for selenium were taken from Bondi et al. [25].
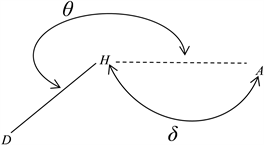
Scheme 3. Geometric parameters of the hydrogen-bonding: interaction distance (d) and hydrogen-bond angle (θ).
![]()
![]()
Table 5. Hydrogen-bonds derived from docking of GK with derivatives as ligands.
All values of the δ distance are less than the sum of van der Waals radii of the interacting atoms and the θ angles are greater than the 120˚ recommended limit [26]. This suggests that the hydrogen bonds were effective. Interestingly, all derivatives like the S41 ligand were able to form two specific hydrogen bonds with the Arg 63 amino acid residue, which is catalytically crucial for therapeutic properties of GK activators [6] [27].
It is noteworthy that beside hydrogen bonds and lipophilic interactions, ligands containing divalent S or Se atoms formed sigma-hole bonds, also known as chalcogen bonds as shown in Figure 6.
![]()
Figure 6. The pattern of chalcogen bondings in the 9Se (Se---O) (a), 3Se (Se----N) (b) and 3S (S---N) (c).
The bond pattern in Figure 6 suggests that the chalcogen interaction were directed along the extensions of covalent bonds of the chalcogen atom as expected [28]. Table 6 reports the distances (δ) and angles (θ) of chalcogen bonds formed between the 3S, 3Se and 9Se ligands and lone pairs of atoms of the amino acid residues acting as bond acceptors in the binding site.
As shown in Table 6, the mean value of 144˚ obtained for the bond angle (θ1) was greater than 140˚ recommended limit for sigma-hole interactions [28]. The δ distances between interacting atoms are less than the sums of their respective
![]()
Table 6. Chalcogen bonds derived from docking of GK with ligands containing S or Se atoms.
van der Waals radii suggesting that the interactions contacts were close contacts. Chalcogen bonds are short, weakly attractive and linear contacts between regions of lower electronic density (σ-hole) of donor atoms and electron lone pairs of nucleophile atoms, acting as sigma-hole acceptors [29]. Directed interactions seem to be characteristic of interactions involving chalcogen atoms. Indeed, Bibelayi et al. (2016) demonstrated that hydrogen bonding to monovalent S and Se atoms in thiourea, thioamide, selenoamide and selenourea derivatives were also nearly linear [30].
4. Conclusion
Investigations of the activation of glucokinase as a protein target in the treatment of diabetes have benefited from evident progress in drug design based on ligand-protein docking and quantitative structure-activity relationship (QSAR). In the present work, the docking of GK with seleno, sulfo and oxo derivatives of the S41 carboxamide activator revealed that all derivatives were bonded to the allosteric site of GK with relatively high affinity. According to ligand binding energies, the main forces stabilizing the complexes were lipophilic interactions enhanced by hydrogen bonds, but also chalcogen bonds in the case of seleno and sulfo derivatives. The increase of thermodynamic stability of these complexes compared with 3S41 can be ascribed to enhancement of the lipophilic character of S41 derivatives. Interestingly, twelve of the S41 derivatives showed stronger binding affinity than the co-crystallized ligand, while maintaining the two hydrogen bonds with crucial Arg63 amino acid residue. Therefore, this study provides an additional support to recent advances in use of the molecular docking as a powerful tool for design and discovery of activators with improved pharmacological properties. The study of the twelve best ranked derivatives should be extended to pharmacophore investigations on their therapeutic properties. The prediction of their activator activities through molecular dynamic simulation and QSAR studies based on ligand chemical and quantum descriptors should also be useful for experimental and clinical trials.
Acknowledgements
Glodi M. Ndefi, Kilembe Thambwe Jason, Bibelayi Dikima Didi and Lundemba Singa Albert are grateful to the Cambridge Crystallographic Data Centre (CCDC) for funding Master and PhD fellowships, and additional financial and material support.