Identification, Synthesis, Isolation and Spectral Characterization of Multidrug-Resistant Tuberculosis (MDR-TB) Related Substances ()
1. Introduction
Tuberculosis (TB) is a chronic infectious disease caused by Mycobacterium tuberculosis. Among acid-fast bacteria, human Mycobacterium tuberculosis has been widely known. It is said that one-third of the human population is infected with this bacterium. Tuberculosis, an airborne lung infection, still remains as a major public health problem worldwide. It is estimated that about 32% of the world population is infected with Tuberculosis bacillus and of those, approximately 8.9 million people develop active Tuberculosis and 1.7 million die annually as a result, as per 2004 figures [1] . Human immunodeficiency virus (HIV) infection has been a major contributing factor in the current resurgence of Tuberculosis [2] [3] . In addition to the human Mycobacterium tuberculosis, Mycobacterium africanus and Mycobacterium bovis have also been known to belong to the Mycobacterium tuberculosis group. The recent emergence of multidrug-resistant (MDR) strains of Mycobacterium tuberculosis that are resistant to two major effective drugs, isonicotinic acid hydrazide (INH) [4] and rifampicin (RFP) [5] , have further complicated the world situation [6] .
Tuberculosis is positioned as the second leading cause of death from a single infectious agent worldwide, with an incidence rate of almost 9 million cases and a fatality rate of 1.4 million in 2011. The World Health Organization (WHO) has estimated that if the present conditions remain unchanged, more than 30 million lives will be claimed by TB, between 2000 and 2020 [7] . The current therapy consists of an intensive phase with four drugs, INH, RFP, pyrazinamide (PZA), 8 and streptomycin (SM) [8] or ethambutol (EB) [9] , administered for 2 months followed by a continuous phase with INH and RFP for 4 months [10] . These bacteria known as Mycobacteria, have a strong pathogenicity to humans. Hepatotoxicity, which is induced by rifampicin, isoniazid, and ethambutol, out of the 5 agents used in combination of first-line treatment, is known as an adverse reaction that is developed most frequently. At the same time, Mycobacterium tuberculosis resistant to antitubercular agents, multi-drug-resistant Mycobacterium tuberculosis, and the likes have been increasing of Mycobacterium tuberculosis, making the treatment more difficult. Notwithstanding the successful implementation of directly observed treatment short course (DOTS) in many countries, high prevalence of multi-drug-resistant (MDR) and extensively drug resistant (XDR) tuberculosis has intensified the urgent need for new anti-tubercular drugs.
Thus, there exists an urgent need for the development of potent new antituberculotic agents with low toxicity profiles that are effective against both drug-susceptible and drug-resistant strains of M. tuberculosis and that can shorten the current duration of therapy [11] .
Recently, a novel chemical entity named as (2R)-2,3-dihydro-2-methyl-6-nitro- 2-[[4-[4-[4-(trifluoromethoxy)phenoxy]-1-piperidinyl]phenoxy]methyl]imidazo[2,1-b]oxazole, a promising anti-TB drug candidate from phenotypic high-content screening (HCS) technology inside infected macrophages, has been reported [12] .
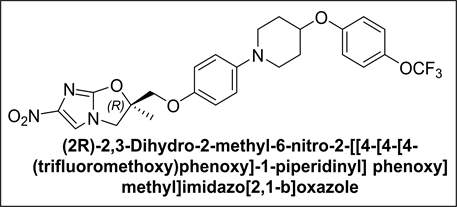
The presence of its related compounds in a drug substance, can have a significant impact on the quality and safety of the drug product. Therefore, it is necessary to study the impurity profile of any active pharmaceutical ingredient (API) and control it during the manufacturing of a drug product. As per the general guidelines recommended by ICH [4] to qualify the drug substance, the amount of acceptable level for a known and unknown related compound should be less than 0.15% and 0.10%, respectively. To meet the stringent regulatory requirements, the impurities present in the drug substance must be identified and characterized thoroughly.
Very recently, an efficient, industrial scale synthesis of (2R)-2,3-dihydro-2- methyl-6-nitro-2-[[4-[4-[4-(trifluoromethoxy)phenoxy]-1-piperidinyl]phenoxy] methyl]imidazo[2,1-b]oxazole has been described. During its asymmetric synthesis, we came across many process related impurities and some of them were captured in our prior art. To comprehend the complete impurity profile of (2R)-2,3-dihydro-2-methyl-6-nitro-2-[[4-[4-[4-(trifluoromethoxy)phenoxy]-1-piperidinyl]phenoxy] methyl]imidazo[2,1-b]oxazole and to compare the extent of contamination of the impurities in it, we decided to synthesize all the possible impurities. In this article, we have reported the synthesis, isolation and spectral characterization of impurities observed during our process development of (2R)-2,3-dihydro-2-methyl-6-nitro-2-[[4-[4-[4-(trifluoromethoxy)-phenoxy]-1-piperidinyl] phenoxy] methyl]imidazo[2,1-b]oxazole. Also, given process describes preparation of (2R)-2,3-dihydro-2-methyl-6-nitro-2-[[4-[4-[4-(trifluoro- methoxy)-phenoxy]-1-piperidinyl]phenoxy]methyl]imidazo[2,1-b]oxazole.
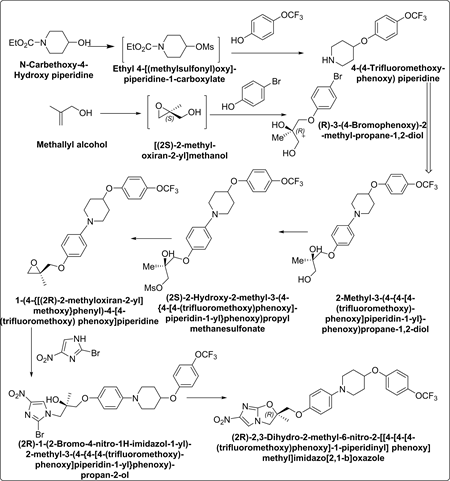
Synthetic Scheme-1:
2. Chemistry
All reactions were carried out under an argon/nitrogen atmosphere in oven-dried glassware with magnetic/mechanical stirring. All reagents and solvents used/mentioned in this study were commercially available. All reactions were monitored by TLC using commercial silica gel plates/HPLC. Analytical thin-layer chromatography (TLC) was performed on 0.25 mm silica gel 60-F254 plates (Merck). Visualization was accomplished with 254 nm UV light and phosphomolybdic acid (PMA) or potassium permanganate staining followed by purification of related substances carried out by flash chromatography on silica gel 60 (Merck, 230 - 400 mesh). NMR spectra were recorded on Bruker 300 MHz Avance NMR spectrometer with tetra methyl silane as an internal reference. Data are reported as ap. = apparent, s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, br. = broad; coupling constant(s) are given in hertz; with integration). Mass spectra were recorded on Waters Xevo G2-XS Q-TOF LC/MS/MS system
Detailed Synthetic Process
Asymmetric synthesis of (R)-3-(4-Bromophenoxy)-2-methyl-propane-1,2- diol:
Synthetic Scheme-2:

Under inert atmosphere, Titanium(IV) isopropoxide was added to a suspension of β-Methallyl alcohol (83.36 g, 1.1560 mol.), (−)-Diisopropyl D-tartrate (16.25 g, 1.1560 mol.) and activated molecular sieves powder* (40 g) in toluene (400 ml) at −12˚C ± 5˚C. After stirring for 30 min., Cumene hydroperoxide (373.86 g, 1.9652 mol.) was added slowly at same temperature and stirring was continued. After completion of reaction (by GC), Dimethyl sulfoxide (90.32 g, 1.1560 mol.) was added slowly and under stirring at <15˚C and stirring was continued for 6 hrs. Thereafter, 15 g of Hyflo was added and reaction mass filtered. To the obtained filtrate, 4-Bromophenol (100 g, 0.5780 mol.) and ~25% w/w aq. sodium hydroxide solution (25.43 g of sodium hydroxide in 77 ml of water) were added sequentially and temperature was raised to 40˚C ± 2˚C. After completion of reaction, which takes ~9 hrs, neutral carbon (2 g) and Hyflo (40 g) were added, stirred and filtered. Obtained filtrate was washed with water and concentrated and crystallised in toluene to yield 120 g (R)-3-(4-Bromophenoxy)-2- methyl-propane-1,2-diol.
Synthesis of 4-(4-Trifluoromethoxyphenoxy) piperidine:
Synthetic Scheme-3:
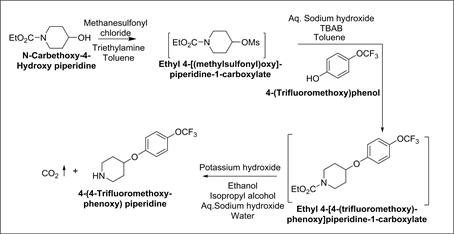
Under inert atmosphere, to a mixture of N-Carbethoxy-4-Hydroxy piperidine (194 g; 1.120 mol.) and methane sulfonyl chloride (141 g, 1.231 mol.) in toluene (500 ml), Triethylamine (148 g, 1.462 mol.) was added at <20˚C and stirred at 20˚C ± 3˚C. After completion of reaction, reaction mass was quenched with water and organic layer was separated. 4-(Trifluoromethoxy)phenol (100 g, 0.561 mol.), aqueous Tetra butyl ammonium bromide solution (62 g in water 62 ml, 0.191 mol.) and aqueous sodium hydroxide solution (87.3 g in 350 ml of water) was added sequentially to the obtained organic layer and temperature was raised to 86˚C ± 3˚C slowly. After completion of reaction, reaction mass was cooled to 20˚C ± 3˚C, organic layer was separated and washed with water. To the obtained organic layer, ethanol (200 ml) and potassium hydroxide (283 g, 5.035 mol.) were added sequentially. Thereafter, reaction mass temperature was raised to 86˚C ± 3˚C slowly. After completion of reaction, reaction mass was concentrated and product was extracted/washed using toluene/water. Thus, obtained organic layer was concentrated and product was crystallized with isopropyl alcohol/water mixture (100 ml/1000ml) and aqueous sodium hydroxide solution (12.5 g in 50 ml water) to provide 140 g of 4-(4-Trifluoromethoxyphenoxy) piperidine.
Synthesis of 1-(4-{[(2R)-2-Methyloxiran-2-yl]methoxy}phenyl)-4-[4- (trifluoromethoxy)-phenoxy]piperidine:
Synthetic Scheme-4:
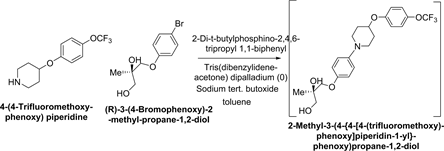
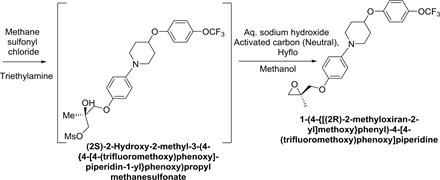
Under inert atmosphere, after charging 4-(4-Trifluoromethoxyphenoxy) piperidine (100 g; 0.3827 mol.), (R)-3-(4-Bromophenoxy)-2-methyl-propane-1,2-diol (105 g, 0.4021 mol.), 2-Di-t-butylphosphino-2,4,6-tripropyl 1,1-biphenyl (0.975 g, 0.0022 mol.), Tris(benzylideneacetone) dipalladium (0) (0.876 g, 0.00095 mol.) and sodium tert. butoxide (42.31 g, 0.44 mol.) sequentially to toluene (300 ml), temperature was raised to 73˚C ± 2˚C. After completion of reaction, reaction mass was cooled to 0˚C ± 3˚C and diluted with toluene (400 ml). Thereafter, triethylamine (77.47 g, 0.766 mol.) and methane sulfonyl chloride (50.43 g, 0.411 mol.) were added sequentially and stirred at this temperature. After completion of reaction, aqueous sodium hydroxide solution (76.75 g of sodium hydroxide in 230 g of water) was added and temperature was raised to 40˚C ± 2˚C and stirred. After completion of reaction, Hyflo (10 g) was added to reaction mass, filtered and washed with toluene. From filtrate, organic layer was separated and washed with aqueous ammonium chloride (60 g of ammonium chloride in 240 g of water). Finally, obtained organic layer was concentrated and crystallised with isopropyl alcohol (650 ml) and water (163 ml) to yield 125 g of 1-(4-{[(2R)-2-methyloxiran-2-yl]methoxy}phenyl)-4-[4-(trifluoromethoxy)phenoxy]piperidine.
Synthesis of (2R)-2-Methyl-6-nitro-2-[(4-{4-[4-(trifluoromethoxy)phenoxy] piperidin-1-yl}phenoxy)methyl]-2,3-dihydroimidazo[2,1-b][1,3]oxazole
Synthetic Scheme-5:

Charge 1-(4-{[(2R)-2-methyloxiran-2-yl]methoxy}phenyl)-4-[4-(trifluoromethoxy) phenoxy]-piperidine (100 g, 0.236 mol.), 2-Bromo-4-nitro imidazole (52 g, 0.272 mol.) and anhydrous sodium acetate (4 g, 0.048 mol.) in tert. butyl acetate (100 ml) and raise the temperature to 98˚C ± 3˚C. After reaction completion, mass was cooled to 27˚C - 30˚C and diluted with methanol (700 ml). Further, reaction temperature was lowered to 2˚C ± 3˚C and added aqueous sodium hydroxide solution slowly (15.78 g in 47.34 g water). Again, temperature was raised to 27˚C - 30˚C and stirred. After reaction completion, reaction mass was diluted with water (150 ml) and with ethyl acetate (50 ml) and temperature raised to 50˚C ± 5˚C. After 1hr. of stirring contents were cooed to 23˚C ± 3˚C, stirred for 1 hr. and filtered and washed with methanol and water. Thus, obtained wet product was finally crystallized with acetone (400 ml) and ethanol (400 ml) to yield 100 g of (2R)-2,3-dihydro-2-methyl-6-nitro-2-[[4-[4-[4-(trifluoromethoxy)phenoxy]-1-piperidinyl]phenoxy]methyl]imidazo[2,1-b] oxazole.
3. Discussion on Related Substances
➢ 1-(Methylsulfonyl)-4-[4-(trifluoromethoxy)-phenoxy] piperidine:
Origin: 4-(4-Trifluoromethoxyphenoxy) piperidine intermediate present in 2-Methyl-3-(4-{4-[4-(trifluoromethoxy)phenoxy]piperidin-1-yl-}phenoxy)propane-1,2-diol, can result in 1-(Methylsulfonyl)-4-[4-(trifluoromethoxy)phenoxy] piperidine during preparation of (2S)-2-Hydroxy-2-methyl-3-(4-{4-[4-(trifluo- romethoxy)phenoxy]piperidin-1-yl}phenoxy)propyl methane sulfonate.
Synthetic Scheme-6:

Synthesis of 1-(Methylsulfonyl)-4-[4-(trifluoromethoxy)-phenoxy] piperidine:
Synthetic Scheme-7:
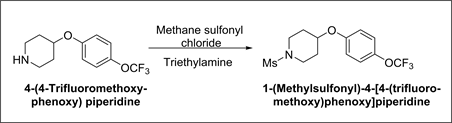
Under inert atmosphere, to a mixture of 4-(4-Trifluoromethoxyphenoxy) piperidine (5 g, 0.0191 mol.), triethylamine (2.51 g, 0.0248 mol.) and methylene chloride (50 ml), methane sulfonyl chloride (2.41 g, 0.0210 mol.) was added at room temperature and stirred for 1 hr. at the same temperature. After reaction completion, reaction mass was quenched with water and organic layer was separated. The organic layer was then washed with water. Organic layer was concentrated and crystallized in ethanol to afford 5 g of 1-(Methylsulfonyl)-4-[4-(trifluoromethoxy)-phenoxy]piperidine.
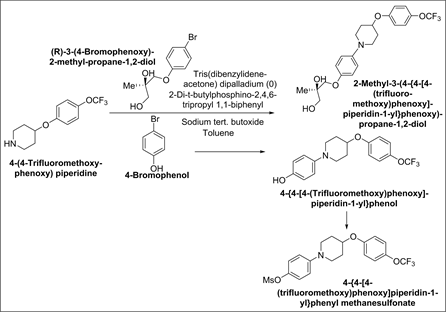
➢ 4-{4-[4-(Trifluoromethoxy)phenoxy]-piperidin-1-yl}phenol and 4-{4-[4- (trifluoromethoxy)phenoxy]piperidin-1-yl}phenyl methanesulfonate
Synthetic Scheme-8:

Origin: Presence of 4-Bromophenol in (R)-3-(4-Bromophenoxy)-2-methyl- propane-1,2-diol intermediate can carry through the synthesis to yield 4-{4-[4-(Trifluoromethoxy)phenoxy]piperidin-1-yl}phenol in 2-Methyl-3-(4-{4- [4-(trifluoromethoxy)phenoxy]piperidin-1-yl}phenoxy)-propane-1,2-diol intermediate preparation and afterward 4-{4-[4-(trifluoromethoxy)phenoxy] piperidin- 1-yl}phenyl methane sulfonate in (2S)-2-Hydroxy-2-methyl-3-(4-{4-[4-(trifluoro- methoxy)phenoxy]piperidin-1-yl}phenoxy)propyl methane sulfonate intermediate.
Synthetic Scheme-9:
Synthesis of 4-{4-[4-(Trifluoromethoxy)phenoxy]-piperidin-1-yl}phenol:
Synthetic Scheme-10:
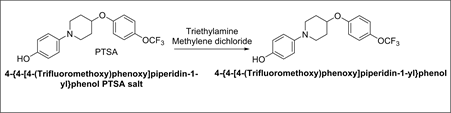
A mixture of 4-{4-[4-(Trifluoromethoxy)phenoxy]piperidin-1-yl}phenol PTSA salt (10 g, 0.019 mol.), methylene chloride (100 ml) and water (100 ml), triethylamine (5 ml) was added and stirred for 30 minutes. Settled and separated the layers. The organic layer was washed with water and concentrated under vacuum to afford 5g of 4-{4-[4-(Trifluoromethoxy)phenoxy]piperidin-1-yl}phenol.
Synthesis of 4-{4-[4-(Trifluoromethoxy)phenoxy]piperidin-1-yl}phenyl methane sulfonate:
Synthetic Scheme-11:

Under inert atmosphere, to a mixture of 4-{4-[4-(Trifluoromethoxy)phenoxy] piperidin-1-yl}phenol (10 g, 0.028 mol.), triethylamine (5.7 g, 0.0566 mol.) and ethyl acetate (100 ml), methane sulfonyl chloride (3.2 g, 0.028 mol.) was added and stirred at room temperature for 1 hr. After completion of reaction, reaction mass was quenched with water and organic layer was separated. The organic layer was concentrated under vacuum to afford 10 g of 4-{4-[4-(Trifluoromethoxy) phenoxy]piperidin-1-yl}phenyl methane sulfonate.
➢ 4-Bromophenyl methane sulfonate

Origin: 4-Bromophenol is being used in (2R)-3-(4-Bromophenoxy)-2-me- thylpropane-1,2-diol Intermediate preparation. During preparation of 1-(4- {[(2R)-2-Methyloxiran-2-yl]methoxy}phenyl)-4-[4-(trifluoromethoxy)phenoxy] piperidine, 4-Bromophenol present in (2R)-3-(4-bromophenoxy)-2-methylpropane- 1,2-diol Intermediate can yield in 4-Bromophenyl methane sulfonate.
Synthesis of 4-Bromophenyl methane sulfonate
Synthetic Scheme-12:
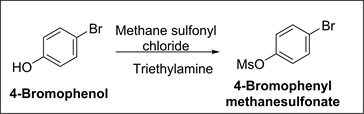
Under inert atmosphere, to a mixture of 4-Bromo phenol (10 g, 0.0578 mol.), triethylamine (8.77 g, 0.0867 mol.) and toluene (50 ml), methane sulfonyl chloride (8.6 g, 0.0751 mol.) was added and stirred at room temperature for 16 hrs. After completion of reaction, reaction mass was quenched with water and organic layer was separated. The organic layer was washed with aq. sodium bicarbonate solution. The organic layer was concentrated to afford 11 g of 4-Bromophenyl methane sulfonate.
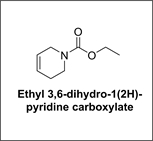
➢ Ethyl 3,6-dihydro-1(2H)-pyridine carboxylate
Origin:
Synthetic Scheme-13:
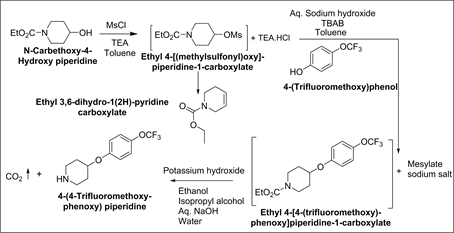
Synthesis of Ethyl 3,6-dihydro-1(2H)-pyridine carboxylate:
Synthetic Scheme-14:

A mixture of ethyl 4-[(methylsulfonyl)oxy]piperidine-1-carboxylate (70 g, 0.2788 mol.), toluene (350 ml) and N,N-dimethyl acetamide (350 ml), potassium tertiary butoxide (38 g, 0.338 mol.) was added and temperature was raised to 50-55˚C and the contents were stirred at the same temperature for 24 hrs. After completion of reaction, reaction mass was quenched with water and organic layer separated. The organic layer was washed with water. The organic layer was concentrated under vacuum to afford 37.5 g of Ethyl 3,6-dihydro-1(2H)-pyridine.
➢ Process related substances of 1-(4-{[(2R)-2-Methyloxiran-2-yl]methoxy} phenyl)-4-[4-(trifluoro-methoxy)phenoxy]piperidine:
![]()
Origin:
Synthetic Scheme-15:
![]()
Synthesis of (2S)-3-(4-Bromophenoxy)-2-hydroxy-2-methylpropyl methane sulfonate:
Synthetic Scheme-16:
![]()
Under inert atmosphere, to a mixture of (R)-3-(4-Bromophenoxy)-2-methyl- propane-1,2-diol (20 g, 0.076 mol.), triethylamine (9.36 g, 0.0919 mol.) and ethyl acetate (150 ml), methane sulfonyl chloride (9.4 g, 0.0820 mol.) was added at 0˚C ± 3˚C and stirred at the same temperature for 1 hr. After completion of reaction, reaction mass was quenched with water and organic layer was separated. The organic layer was washed with water. The organic layer was concentrated to afford crude residue which was further isolated in hexanes to afford 23 g of (2S)-3-(4-Bromophenoxy)-2-hydroxy-2-methylpropyl methane sulfonate.
Synthesis of (2S)-3-(4-Bromophenoxy)-2-methylpropane-1,2-diyl dimethanesulfonate:
Synthetic Scheme-17:
![]()
Under inert atmosphere, to a mixture of (R)-3-(4-Bromophenoxy)-2-methyl- propane-1,2-diol (10 g, 0.0384 mol.), triethylamine (11.6 g, 0.1146 mol.) and ethyl acetate (100 ml), methane sulfonyl chloride (11 g, 0.0960 mol.) was added below 20˚C and stirred at room temperature for 1 hr. After completion of reaction, reaction mass was quenched with water and organic layer was separated. The organic layer was washed with water. The organic layer was concentrated to afford crude residue which was further purified by column chromatography using silica gel, eluent ethyl acetate: hexanes (1:1) to afford 5g of (2S)-3-(4-Bro- mophenoxy)-2-methylpropane-1,2-diyl dimethane sulfonate.
Synthesis of (2S)-2-Methyl-3-(4-{4-[4-(trifluoromethoxy)phenoxy]piperidin- 1-yl}phenoxy)-propane-1,2-diyl dimethane sulfonate
Synthetic Scheme-18:
![]()
Under inert atmosphere, to a mixture of 2-methyl-3-(4-{4-[4-(trifluoromethoxy) phenoxy]piperidin-1-yl}phenoxy)propane-1,2-diol (15 g, 0.034 mol.), triethylamine (13.76 g, 0.136 mol.) and ethyl acetate (150 ml), methane sulfonyl chloride (8.92 g, 0.085 mol.) was added at 0˚C ± 3˚C and stirred at room temperature for 1 hr. After completion of reaction, reaction mass was quenched with water and organic layer was separated. The organic layer was washed with water. The organic layer was concentrated under vacuum to afford 18g of (2S)-2-methyl-3- (4-{4-[4-(trifluoromethoxy) phenoxy]piperidin-1-yl}phenoxy)propane-1,2-diyl.
➢ (S)-3-(4-Bromophenoxy)-2-methyl-propane-1,2-diol and corresponding anti-TB drug candidate Enantiomer
![]()
Origin:
Synthetic Scheme-19:
![]()
Synthesis of (2S)-3-(4-Bromophenoxy)-2-methylpropane-1,2-diol:
Synthetic Scheme-20:
![]()
A mixture of beta-Methallyl alcohol (83.36 g, 1.1560 mol.), (+) Diisopropyl L-tartrate (16.25 g, 0.0694), Molecular Sieves powder (Grade 4A˚) (40.00 g), and toluene (400 ml) was stirred at −17˚C to −15˚C. Titanium tetra isopropoxide (16.43 g, 0.0578 mol.) was added to the mixture followed by stirring at −18˚C to −16˚C for 30 minutes. Thereafter, 80% Cumene hydroperoxide (373.86 g, 1.9652 mol.), was added thereto dropwise at −18˚C to −16˚C over a period of 2 hours, followed by stirring at −11 to −9˚ for 5 hours. Dimethyl sulfoxide (90.32 g, 1.1560 mol.) was added thereto at below 15˚C over a period of 30 minutes. The resulting mixture was stirred at room temperature for 6 hours. After adding hyflo (15 g), continued stirring for 30 minutes. The reaction mixture was subjected to filtration, obtained a toluene solution of (S)-2-methylglycidyl alcohol.
4-Bromo phenol (100 g, 0.5780 mol.), and a 25% w/w sodium hydroxide aqueous solution (101.72 g) were added to the toluene solution thus obtained, followed by stirring at 40˚C for 9 hours. After cooling the reaction mixture to room temperature, activated carbon (2 g) and hyflo (40 g) were added thereto. The reaction mixture was then stirred for 30 minutes and subjected to filtration. The toluene layer was washed with water (500 ml), and then the toluene and cumyl alcohol were distilled off under reduced pressure. Toluene was added to the concentrated residue followed by heating to 70˚C to dissolve the concentrated residue. The resulting solution was cooled to room temperature (24˚C to 26˚C) and continued stirring for 6 hours. The precipitated product was collected by filtration and then washed with toluene (100 ml). The product was dried under vacuum for 8 hours at 50˚C - 55˚C to obtain 92 g of the (S)-3-(4-Bromo- phenoxy)-2-methyl-propane-1,2-diol.
(2S)-2,3-dihydro-2-methyl-6-nitro-2-[[4-[4-[4-(trifluoromethoxy)phenoxy]-1-piperidinyl] phenoxy] methyl]imidazo[2,1-b]oxazole:
This is prepared by following the similar procedure given for the preparation of title compound i.e. anti-TB drug candidate by using (S)-3-(4-Bromophenoxy)- 2-methyl-propane-1,2-diol instead of (R)-3-(4-Bromophenoxy)-2-methyl-propane- 1,2-diol.
➢ (2R)-2-[(4-Bromophenoxy)methyl]-2-methyloxirane and (2R)-2-[(4- bromophenoxy)methyl]-2-methyl-6-nitro-2,3-dihydroimidazo[2,1-b][1,3] oxazole
![]()
Origin: Presence of 3-(4-bromophenoxy)-2-methylpropane-1,2-diol in 2-methyl- 3-(4-{4-[4-(trifluoromethoxy)phenoxy]piperidin-1-yl}phenoxy)propane-1,2-diol intermediate, can carry through the synthesis to yield (2R)-2-[(4-bromophenoxy) methyl]-2-methyloxirane and (2R)-2-[(4-bromophenoxy)methyl]-2-methyl- 6-nitro-2,3-dihydroimidazo[2,1-b][1,3] oxazole impurities.
Synthesis of (2R)-2-[(4-Bromophenoxy)methyl]-2-methyloxirane:
Synthetic Scheme-21:
![]()
A mixture of (2S)-3-(4-Bromophenoxy)-2-hydroxy-2-methylpropyl methane sulfonate (10 g, 0.0194 mol.) and methanol (150 ml), potassium carbonate (4.2 g, 0.0380 mol.) was added at 0˚C ± 3˚C and raised the temperature to room temperature and stirred the contents for 1 hr. After completion of reaction, the reaction mixture was concentrated under vacuum. Charged ethyl acetate and water. Stirred, settled and separated the layers. The organic layer was separated and washed with water. The organic layer was concentrated under vacuum to afford 5g of (2R)-2-[(4-Bromophenoxy)methyl]-2-methyloxirane.
Synthesis of (2R)-2-[(4-Bromophenoxy)methyl]-2-methyl-6-nitro-2,3- dihydroimidazo[2,1-b][1,3]oxazole:
Synthetic Scheme-22:
![]()
To a mixture of 3-(4-Bromophenoxy)-2-methylpropane-1,2-diol (25 g, 0.0958 mol.), triethylamine (19.3 g, 0.1901 mol.) and ethyl acetate (250 ml), methane sulfonyl chloride (12.55 g, 0.1095 mol.) was added at 0˚C ± 3˚C and stirred at the same temperature. After completion of reaction, reaction mass was quenched with water and organic layer was separated. The organic layer was washed with water. The organic layer was concentrated under vacuum to afford residue. To the obtained residue was added methanol (300 ml) and potassium carbonate (26.4 g, 0.191 mol.) at 0˚C ± 3˚C and stirred at the same temperature for 1 hr. After completion of reaction, the reaction mass was concentrated under vacuum to remove methanol. The residue thus obtained dissolved in toluene and washed with water. The organic layer was concentrated under vacuum to obtained residue. To the obtained residue was added 2-Chloro-4-nitro imidazole (8 g, 0.0544 mol.) and anhydrous sodium acetate (0.89 g, 0.0108 mol.) in t-Butyl acetate (20 ml) and raise the temperature to 98˚C ± 3˚C and stirred the contents for 1 hr. After reaction completion, mass was cooled to 27˚C - 30˚C and diluted with methanol (140 ml). Further, reaction temperature was lowered to 2˚C ± 3˚C and added aqueous sodium hydroxide solution slowly (15.57 g). Again, temperature was raised to 27˚C - 30˚C and stirred. After reaction completion, reaction mass was diluted with water (30 ml) and with ethyl acetate (30 ml) and temperature raised to 50˚C ± 5˚C. After 1hr. of stirring contents were cooed to 23˚C ± 3˚C, stirred for 1 hr. and filtered and washed with methanol and water and dried the product under vacuum to yield 14.5 g of (2R)-2-[(4-bromophenoxy) methyl]-2-methyl-6-nitro-2,3-dihydroimidazo[2,1-b][1,3]oxazole .
4. Conclusion
In summary, related substances plausible to be contemporary in (2R)-2,3-dihydro- 2-methyl-6-nitro-2-[[4-[4-[4-(trifluoromethoxy)phenoxy]-1-piperidinyl]phenoxy]methyl]imidazo[2,1-b]oxazole drug substance were acknowledged, synthesized and extensively characterized. Also, plausible origin for these related substances has been provided to have a better control for these and make the product free from these.
Acknowledgements
Our group would like to thank the Department of Scientific and Industrial Research India, Dr. Hari Babu (COO Mylan), Sanjeev Sethi (Chief Scientific Office Mylan Inc.); Dr Abhijit Deshmukh (Head of Global OSD Scientific Affairs); Dr Yasir Rawjee {Head-Global API (Active Pharmaceutical Ingredients)}, Dr Sureshbabu Jayachandra (Head of Chemical Research) Dr. Suryanarayana Mulukutla (Head Analytical Dept MLL API R & D) as well as analytical development team of Mylan Laboratories Limited for their encouragement and support. We would also like to thank Dr Narahari Ambati (AGC-India IP) & his Intellectual property team for their support.