Surface Species Formed during Methane Oxidation over Some Rare Earth Elements Oxides ()
1. Introduction
Due to the noticeable decrease of oil reserves and high oil prices globally, and protecting the Earth from the gaseous pollutants and global warming, attention has been directed to search for alternatives to oil, primarily natural gas (methane) to produce chemicals and fuels cleaner and more environmental-friendly and economic value added, to maintain global economic growth and fuel supplies, through research and development programs designed to convert methane to hydrocarbons, an economically feasible ways that directly or indirectly as a process of oxidative coupling of methane (OCM) [1] - [6] .
It is industrially important to convert alkanes C1-C4 to oxygenated compounds, which can be used as raw materials for industries. Despite the fact that these transformations favored heat, the problem of industries lies in finding the appropriate operational conditions to generate intermediates containing active and strong C-H or C-C bonds, compared to oxidation of the alkanes to carbon dioxide CO2 and water H2O.
The oxidative coupling of methane (OCM) is one of the most important reactions in relation to chemical utilization of natural gas. Since pioneering works of Keller et al. [7] , many studies have been made to find an efficient catalyst. In this area, catalysts based upon lanthanide oxide have been shown to have significance, but as yet largely unrealized potential for catalyzing the oxidation of methane [8] - [17] .
However, it has been found in a series of catalytic oxidation that the catalytic activity of the lanthanide oxides significantly differs from each other. Sazonov et al. [18] compared the catalytic activity in the oxidation of hydrogen and propylene with that of the isotopic exchange of oxygen, and suggested that the catalytic activity depends on the binding energy of oxygen to the surface and on the valance of the lanthanide ions. Others suggested that the catalytic activity of the lanthanide oxides depends on the electronic configuration of the inner 4f subshell [19] .
Considerable progress has been made in understanding the mode by which methane is activated for the oxidative addition to metal centers and for the generation of methyl radicals CH3•, that are formed at the surface of the catalyst [20] . Which eventually changes at high temperature to both oxygen compounds (methanol and formaldehyde), and to higher hydrocarbons (mainly ethane, ethylene) [21] [22] [23] [24] .
Wang et al. [25] reported that methane could be transformed to benzene on transition metal ions supported by HZSM-5 catalysts under non-oxidative condition. This study as Keller et al. and Driscoll et al. [7] [26] proposed a mechanism for the oxidative coupling of CH4 over metal oxides, in which methyl radicals CH3• formed into the gas phase through a surface reaction coupled on the surface to form ethane. While the formation of the surface-generated methyl radicals is an accepted part of both mechanisms, it remains to be demonstrated whether subsequent surface reactions or gas-phase reactions of the methyl radicals constitute the major pathway for the oxidative coupling of methane.
Wu et al. [27] , suggested that, Lanthana, gave a methane conversion with a high selection toward C2+, and an oxide interaction provided the formation of additional oxygen vacancies which exhibited higher selectivity to C2H4. Several papers [28] [29] [30] , examined the abilities of the lanthanide oxides to catalyze the oxidative coupling of CH4 to form C2H6 and C2H4 Scheme 1.
There is no conclusive evidence of any duplication of methyl free radicals on the surface of heterogeneous or heterogeneous catalysts that lead to C2H6 and C2H4, but it is expected that this may be due to the formation of methyl free radicals in the gas phase on the catalyst surface, later mating C2H6, or enter into a series of successive surface reactions that eventually lead to full oxidation [31] [32] [33] [34] .
Methyl radicals react extensively with the lanthanide-metal oxides CeO2, Pr6O11, and Tb4O7 which have multiple cationic oxidation states [35] [36] . By contrast, the oxides La2O3, Nd2O3, Sm2O3, Eu2O3, and Yb2O3 react with CH3• radicals to only a small extent. Consistent with this observation, the former group of oxides is ineffective in the generation of CH3• radicals which emanate into the gas phase.
Catalytic studies with a series of rare earth oxides have been undertaken to observe the effect of periodic trends in basicity and cationic 4f-electron configuration. It is known that high surface basicity of catalyst is necessary to enhance the C2 selectivity and improve the catalytic performance of the metal oxides, for oxidative coupling of methane (OCM) [37] - [42] . The catalytic activity of lanthanide oxides for oxidative coupling of methane showed maxima at Sm, Gd, and Ho oxides. A different activity pattern was ascribed to the difference in the sample source and in the pretreatment of samples [43] .
The importance of the specific surface area of the catalyst was pointed out in oxidative coupling of methane. Tagawa et al. [44] demonstrated that, an amorphous LnAlO3 catalyst shows a high activity for the C2 hydrocarbon formation. The active sites were concluded to be formed on the surface of an amorphous phase, because the catalytic activity decreased with the growth of the crystalline perovskite phase [45] .
The C2 hydrocarbon formation rate is proportional to the increase in lanthanide ionization potentials. This suggests that the basicity of the lanthanide, or the tendency to lose anions, is closely related to the formation of the active sites on the catalyst surface [46] [47] [48] [49] . Temperature-programmed adsorption studies on a series of catalysts indicated that oxygen molecules were
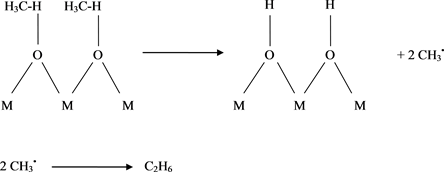
Scheme 1. Surface generated gas-phase CH3• radical.
desorbed from the catalyst during the heating with reaction temperature, and two kinds of oxygen adsorption sites were characteristic of the active amorphous catalysts, Which is thought to be responsible for the coupling reaction [50] .
In the oxidative coupling of methane reaction catalyzed by metal oxide, the transfer of oxygen to the surface from bulk or gas phase is very important in its catalytic activity, because the abstraction of hydrogen from methane is caused by an oxygen ion present on the surface of metal oxide catalyst [51] [52] [53] . However, an OCM catalyst acts not only as the radical initiator but also as the radical quencher.
In this study, we will try to identify the species formed on the surface of the catalyst during the coupling reaction, leading to products with added economic value.
2. Experimental Methods
2.1. Materials and Catalyst Preparation
2.1.1. Materials
Main materials used in the experiment were of high purity:
・ Methane gas CH4 (99.95%) Air products England.
・ Praseodymium Chloride (99.9%).
・ Europium Chloride (99.9%).
・ Ytterbium Nitrate (99.9%) (Oxidized).
・ Distilled water.
・ Ammonium solution (sp. gr. 0.91 gm/ml, about 25% NH3) BDH Analar BDH chemicals ltd. Poole, England.
・ Hydrochloric acid (sp. gr. 1.18 gm/ml, about 36% HCl) BDH Laboratory Reagents.
2.1.2. Equipment
・ Thermolyne (a subsidiary of Syron) Type 1500 Furnace.
・ Disc maker (Perkin Elmer) Pressurized up to 15 Tons/cm2.
・ F.T.I.R Spectrophotometer (Perkin Elmer) Model 1600.
・ Vacuum line.
Oxides of transition metals (rare earth) would be prepared as described in the literature in pure forms. However, mixed oxides would be prepared by co-precipitation of the appropriate mixture of metal chlorides or nitrates by alkaline solutions. This method (co-precipitation) is often used to prepare oxidic or sulfidic catalysts. A solution of a metal salt (often the nitrate) is combined with an ammonium salt of a suitable anion to precipitate the metal ion as hydroxide, sulfide, oxalate, carbonate… etc. Upon heating to high temperatures (calcinations) the compounds are decomposed to give correspondent metal oxides. Simultaneous addition of the metal salt and the precipitating agent to the precipitation flask gives a more uniform catalyst composition than the sequential addition of the components [54] .
Accordingly, about 5 grams of chlorides or nitrates of lanthanide (rare earth) metals were accurately weighed, and dissolved in a minimal volume of distilled water. Few drops of HCl (1N, pH = 6 - 6.5) were added to the chloride solution, which was slightly cloudy, in order to render such solution completely clear. Lanthanide hydroxides were precipitated by addition of a slightly stoichiometric excess of a dilute aqueous ammonia solution or sodium hydroxide. The resulting gelatinous suspension was allowed 24 hours for coagulation and settling of the precipitate. Before filtration of the resulting precipitate, the mixture was heated to boiling for one hour to change the form of precipitate from gelatinous to a finely powdered granular form.
Filtration was performed on a Buchner funnel (No. 5) under vacuum for 20 hrs, the filter cake was washed with boiling distilled water (5 × 5 ml), collected and dried in an oven at 600˚C for 20 hrs.
Ultimately, lanthanide oxides were obtained and the brittle Praseodymium oxide color was changed from pale green to dark brown. Remaining oxides were fluffy porous and changed from white to light yellow. Yield: 3 grams (60%).
A sample of the metal oxide (0.025 gm) was thoroughly milled into a fine powder with 10 times its weight of KBr. The resultant powder was pressed into a disc using a disc maker, and a pressure equivalent to 10 ton load.
The resulting disc was exposed to a gas stream of hydrocarbons (e.g. methane gas) (22 × 10 cm3/min, 1.2 atoms), with intervals of one hour each at a thermal gradient starting from ambient temperature to about 185˚C - 195˚C, 200˚C - 215˚C and 250˚C - 260˚C, temperature control was done manually.
The infrared spectrum of the lanthanide oxide disc was measured before changing the temperature, to elucidate any reaction that could occur, between the metal oxide and methane, during the flow of methane over the metal oxide (catalyst), as follows:
2.1.3. At 25˚C (Ambient Temperature)
The disc was heated to 110˚C to evaporate any water vapor that may be present on the surface of the disc. After cooling to ambient temperature the infrared spectrum was measured, to ensure the absence of water or any other adsorbed substances on the surface of the disc. Methane gas was passed over the sample for one hour, and the infrared spectrum of the disc was measured (by F.T.I.R. Spectrophotometer (Perkin Elmer) model 1600). The spectrum showed that the disc contained new species. A sample of the expected species (e.g. phenol) was grinded with the same weight of the metal oxide and potassium bromide and measurement of the infrared spectrum was repeated. The two spectra were compared and both proved to contain the same species indicating that the reaction product consisted of phenol.
2.1.4. At Other Temperatures
The reaction disc was heated to a temperature higher than the boiling point of the expected species (t > 185˚C), grinded, pressed under the same pressure and the spectrum was remeasured to assure the absence of any reaction species. The gas was passed over the disc for one hour, at 185˚C - 195˚C, and after cooling at room temperature, the I.R. spectrum was measured. The process was repeated at 200˚C - 215˚C and 250˚C - 260˚C and the resulting spectra showed that at temperature lesser than the boiling point (t > 185˚C) of expected species the same products were produced.
3. Results and Discussion
In a separate set of experiments, methane has been adsorbed on the surface of the three oxides (Pr2O3, Eu2O3, Yb2O3) at room temperature, and at elevated temperatures respectively, at a pressure of ca 1.2 atm. for one hour. The F.T.I.R spectra of each disc have been recorded after evacuation at room temperature. The spectra are presented in Figures 1-4.
A close investigation of the spectra indicates that the three oxides are relatively similar in behavior under the specified comparable conditions. Hence, anyone could be used as a prototype of the other two oxides. This would give a comparative study that could be used as the basis of a generalized phenomenon.
The infrared spectrum region between 4000 and 200 cm−1 can roughly be divided into deferent regions:
1) The X-H stretch region (4000 - 2500 cm−1), where strong contributions of OH, NH, CH and SH stretch vibrations are observed.
2) The triple bond region (2500 - 2000 cm−1), where contributions from gas phase CO (2143 cm−1) and linearly adsorbed CO (2000 - 2200 cm−1) are seen.
3) The double region (2000 - 1500 cm−1), where in catalytic studies bridge
![]()
Figure 1. Infrared absorption spectra of surface species, between 4000 and 200 cm−1 formed by adsorption of methane over Pr2O3, Eu2O3 and Yb2O3 at Room Temperature. These have been prepared by precipitation, and then calcined at 600˚C to get M2O3. Methane was then passed through a disk in KBr. at room temperature, and at elevated temperatures respectively, at a pressure of ca 1.2 atm. and for one hour.
![]()
Figure 2. Infrared absorption spectra of surface species, between 4000 and 200 cm−1 formed by adsorption of methane over Pr2O3, Eu2O3 and Yb2O3 at 185˚C - 195˚C. These have been prepared by precipitation, and then calcined at 600˚C to get M2O3. Methane was then passed through a disk in KBr. at room temperature, and at elevated temperatures respectively, at a pressure of ca 1.2 atm. and for one hour.
![]()
Figure 3. Infrared absorption spectra of surface species, between 4000 and 200 cm−1 formed by adsorption of methane over Pr2O3, Eu2O3 and Yb2O3 at 200˚C - 215˚C. These have been prepared by precipitation, and then calcined at 600˚C to get M2O3. Methane was then passed through a disk in KBr. at room temperature, and at elevated temperatures respectively, at a pressure of ca 1.2 atm for one hour.
bonded CO, as well as carbonyl groups in adsorbed molecules (around 1700 cm−1).
4) The fingerprint region (1500 - 500 cm−1), where all single bonds between carbon and elements such as nitrogen, oxygen, sulphur and halogens are adsorbed.
5) The M-X or metal-adsorbate region (around 450 - 200 cm−1), where the metal-carbon, metal-oxygen and metal-nitrogen stretch frequencies in the spectra of adsorbed species are observed.
![]()
Figure 4. Infrared absorption spectra of surface species, between 4000 and 200 cm−1 formed by adsorption of methane over Pr2O3, Eu2O3 and Yb2O3 at 250˚C - 260˚C. These have been prepared by precipitation, and then calcined at 600˚C to get M2O3. Methane was then passed through a disk in KBr. at room temperature, and at elevated temperatures respectively, at a pressure of ca 1.2 atm. for one hour.
3.1. 4000 - 3000 cm−1 Region
In the segment 4000 - 3000 cm−1, several infrared bands show up. Mainly, we can see the following bands:
Free (OH) group absorbed at 3750 cm−1, while with bonded (OH) group, vibrates at 3650 cm−1. However, to distinguish between the (OH) group and (H2O) molecule adsorbed on the surface of the oxide, we should look at the 3880 - 3300 cm−1 region; where (OH) stretch of water absorbs at 3450 cm−1 and shows marked evidence of hydrogen bonding [55] [56] .
Methane adsorbed on Al2O3 shows the following bands in the region under consideration [57] , 3785, 3740, 3710 cm−1 and it is indicative that the band at 3785 cm−1 is due to hydrogen bonding. However, it is worthy to mention that band at 3750 cm−1 (width of 15 cm−1) is due to (O-H) stretch of the free hydroxyl group and the lower shift is due to hydrogen bonding. For example, in silica we get a band at 3770 cm−1 assigned to free (O-H) and a broadband at 3450 cm−1 due to hydrogen bonding.
In alkylation of hydroxylated surfaces the band at 3750 cm−1 decreases and bands at 2965 and 2862 cm−1 increases. The shift in the 3750 cm−1 band due to the hydrogen bonding is exemplified by the following two examples (Table 1).
The infrared spectra of all the oxides show, invariably, and at all elevated temperatures, sharp adsorption bands at: 3882, 3835, 3741, 3612, 3448 cm−1, which according to our above discussion is a conclusive evidence of the presence of free and hydrogen bonded hydroxyl groups. The band at 3448 cm−1 is assignable to (OH) stretch of water and also shows marked evidence of hydrogen bonding. Hence, one could envisage the following mechanism:
1) Formation of H-bonded hydroxyl group on the surface of the oxide (Scheme 2).
![]()
Table 1. The shift in the 3750 cm−1 band due to the hydrogen bonding.
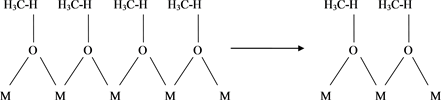
Scheme 2. Formation of H-bonded hydroxyl group on the surface of the oxide.
2) Formation of a free hydroxyl group and CH3• radicals (Scheme 3).
3) Formation of free and hydrogen bonded water (Scheme 4 and Scheme 5).
In the same infrared spectral region of 4000 - 3000 cm−1, we expected to see the following stretching frequencies, - CH2 at 3030 cm−1, ≡ CH at 3300 cm−1. However, such bands have not been observed and hence have been excluded as part of the reaction products under these conditions of reaction.
When we consider the 3000 - 2500 cm−1 region we find the following possible absorptions: The (-CH3) group give rise to a symmetrical (C-H) stretch invariably falling between 2850 - 2890 cm−1, and an asymmetrical stretching frequency at 2940 - 2980 cm−1 [56] . On the other hand the (-CH2-) group gives rise to a band at 2930 cm−1 assigned to the asymmetrical (C-H) stretch and another at 2860 cm−1 assigned to the symmetrical stretch [58] .
The results shown by different oxides exhibit bands at 2965, 2961 and 2855 cm−1 [59] . These have been assigned to an alkoxy group formation as indicated by the mechanism shown by Scheme 5. The specifications of such an alkoxy group will be given later in the discussion.
3.2. 2500 - 2000 cm−1 Region
The infrared absorption bands anticipated at the 2500 - 2000 cm−1 region are:
・ The (O=C=O) asymmetrical stretch at ~2330 cm−1 [60] ,
・ The (-C≡C-) stretch,
o Terminal (-C≡C-) shows weak band 2140 - 2050 cm−1.
o Non-terminal shows at 2300 - 2050 cm−1.
o An unsymmetrical disubstituted acetylenes exhibit infrared band at 2260 - 2190 cm−1 variable intensity.
・ The stretching bands of (-C≡C-) are absent in symmetrically disubstituted acetylenes (alkynes) or acetylene.
・ The intensity of the (-C≡C-) stretching bands is increased by conjugation with a carbonyl group.
The infrared spectra of the absorbed methane show strong I.R. absorption

Scheme 3. Formation of free hydroxyl group.
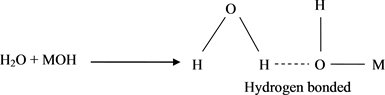
Scheme 4. Formation of hydrogen bonded.
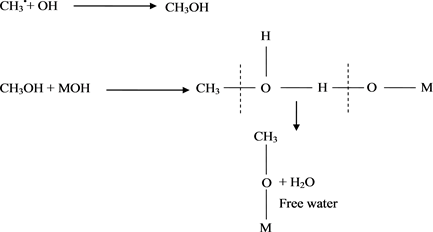
Scheme 5. Formation of free water.
bands at 2355 and 2331 cm−1 which have been assigned to an asymmetrically disubstituted alkyne conjugated to a carbonyl group. Doubling the frequency is an indication of conjugated (-C=C-) groups [61] [62] [63] .
The mechanism of formation of the alkyne entity is envisaged to take place through ethylene formation as is represented by the following:
1) First possibility is the dissociation of the alkoxy group into (CO2) and (CO) which in turn form surface carbon (Cs) and oxygen leading to the formation of the acetylenic surface species [64] (Scheme 6).
2) Moreover, the ≡C-H vibration stretch at 3000 cm−1 region has not been observed which rule out such a schematic mechanism.
Iii formation of ethylene according to the following mechanism [65] [66] (Scheme 7).
Ethylene could also be formed by the reaction of (CH3•) radicals with the surface to form carbon radicals which then combines to yield (C2H4). The latter is

Scheme 6. Dissociation of alkoxy group.
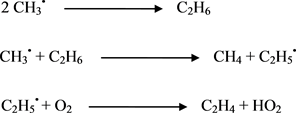
Scheme 7. Formation of ethylene.
dehydrogenated to form an alkyl acetylide surface species, e.g. (Scheme 8).
It is worth noting that ethylene adsorbs irreversibly on clear platinum with dissociation into an acetylenic species and mobile hydrogen [67] .
Also, ethylene is reported to dehydrogenate upon adsorption on platinum (III) surface, transforming into a stable acetylenic structure [68] . Further, structural evidence has been reported for the formation of the ethyldyne group on platinum (III) [69] .
3.3. 2000 - 1800 cm−1 Region
In the 2000 - 1800 cm−1 spectral region, very weak absorption at 1989, 1942, 1919, 1889, 1866, and 1830 cm−1 was observed. In this area, combination and overtone bands appear. Their pattern is characteristic of the substitution pattern of the ring, e.g. indication of the monosubstituted ring.
However, no assignments have been attempted and the bands could be considered either as:
1) Combination and overtones,
or
2) Noise
3.4. 1800 - 1100 cm−1 Region
The 1800 - 1100 cm−1 region of the spectra is a wide range and within its range many absorption bands of different compounds and structures appear, e.g. oxalates, carbonates, nitrates, aldehydes, ketones, alcohols and aromatic hydrocarbons, etc.
Table 2 presents the infrared absorption bands in the spectral region concerned with tentative group assignments.

Scheme 8. Dehydrogenation of C2H4.
![]()
Table 2. I.R. absorption bands and tentative assignments.
The I.R. band at 1742 cm−1 has been tentatively assigned to υ(C=O) stretch and the 1402 cm−1 band is assigned to the CH2 scissors mode of the methylene group. The 1337 cm−1 and 1313 cm−1 bands are due to CH2 deformation. The bands at 1460 and 1390 cm−1 have been tentatively assigned to δas (CH3) and δsy (CH3) of a t-butyl group [56] [70] [71] [72] .
3.5. Below 1100 cm−1 Region
In the region of the spectra below 1100 cm−1 several weak bands appear, mainly the band at 667 and 508 cm−1. The former band at 667 cm−1 could be assigned to chemisorbed CO2, while the later band at 508 cm−1 might tentatively be assigned to [Ln -O-] stretch.
Upon evacuation at higher temperatures, the υstr(-C≡C-) and 667 cm−1 absorption bands diminish, while 2965, 2961, 2855 cm−1 increases, and the (OH) stretching vibration at ~3450 cm−1 builds up. The 1740 cm−1 disappears and the strong bands in the 1550 - 1000 cm−1 appear.
The complement of such results could be integrated and interpreted in terms of formation of several independent species resulting from the breakdown of the originally formed surface species.
Evidence of ring formation has been found in the 667 and 880 cm−1 bands which are assigned to out of plane (C-H) band in the mono-substituted benzene ring, while the bands in the 1000 cm−1 region could be assigned to (C-H) in the plane band. However, the (-C≡C-) stretches could be assigned to the bands at 1513, 1537 and 1551 cm−1.
Mono-substitution of the benzene ring (formation of aromatic compounds) is further indicated by the 1800 - 2000 cm−1 weak absorption bands.
A methylene group could be seen, and the band at 1402 cm−1 is tentatively assigned to the (CH2) scissors mode of the methylene group, while the 1337 cm−1 and the 1313 cm−1 bands are due to (CH2) deformation.
4. Conclusion
This study showed that the oxidative Coupling of methane (OCM) occurs through homogeneous and heterogeneous catalysis reactions that begin by extracting hydrogen of methane in gas phase by activated oxygen on the surface of the catalyst and get methyl radical CH3•. On the other hand (together with H2O co-production), mainly for the production of ethylene C2H6, and the formation of ethylene C2H4 through both heterogeneous and heterogeneous reactions, while Oxygenated carbon products from COX (i.e., CO2 and CO) consist mainly of oxidation of those gaseous phases of hydrocarbons on the catalyst surface. For all the catalysts the conversion increases relatively with increasing the reaction temperature.
Acknowledgements
The author is much grateful to King Abdulalziz City for Science and Technology (KACST), Riyadh, Saudi Arabia for the valuable and continues scientific and moral support.