Validation and Comparison of Two Calibration Methods for the Measurement of Stable Iodine in the Urinary Matrix by ICP-MS: Standard Addition vs. External Calibration ()
1. Introduction
Among the halogen elements, iodine (I) is composed of thirty-seven isotopes among which only one is stable (127I). Although its low quantity within the organism [1] , I is a key micronutrient for the proper functioning of the human body. It is a major component in the synthesis of thyroid hormones especially tetraiodothyronine (thyroxine or T4) and triiodothyronine (T3) and thus is involved in many systems [2] . Among them, it plays a role on the intellectual development by participating in the brain maturation and also contributes to the regulation of many tissue functions (cardiac, muscle or digestive). Thus, the thyroid is a vital organ which may be the site of various disorders such as dysthyroidism (hypothyroidism or hyperthyroidism) or cancerous endpoints meaningfully affecting the health status. In this regard, these major disturbances are greatly feared particularly following a nuclear reactor accident.
Indeed, when such accident occurs, a substantial fraction of releases is mainly composed of iodine isotopes, particularly 131I with a physical half-life of eight days. Once ingested or inhaled, these radionuclides will bind on the thyroid gland where radioactive and non-radioactive iodine are not distinguished and may cause radiation injuries such as thyroid cancer or other thyroid diseases [3] . As an example, in the afterwards of the Chernobyl accident in 1986, a significant number of children at the time of the accident develops thyroid cancer primarily due to the exposure to radioiodines discharged from the plume [4] . As a protective measure against the development of childhood thyroid cancer, French health authorities as many other national organizations elsewhere had defined plans aiming to predistribute stable iodine tablets to people living in the vicinity of nuclear power plants and to organize larger geographical distribution in case of an nuclear accident occurrence. The objective of such countermeasure is to prevent the accumulation of radioactive iodine in the thyroid by administering a single dose of potassium iodide tablets (renewable once whether the evacuation of populations is impossible) to saturate the iodine binding sites [5] . To be effective, this drug must be ideally taken one hour before exposure [6] . However, this administration scheme does not consider the situation of repeated or prolonged releases as seen during the Fukushima Dai-ichi catastrophe. This point clearly represents a gap in the healthcare policies and therefore a major issue of concern for public health authorities [7] [8] .
To interpret properly new experimental data gathered from animal testing that would serve to a significant revision of the iodine doctrine, it was necessary to design a reliable analysis method for the determination of iodine urinary excretion [9] . Various analytical methods have been already developed and are based particularly on the following techniques: instrumental neutron activation analysis [10] , As-Ce catalytic spectrophotometry [11] , ion selective electrodes [12] , electrochemical detection [13] , size chromatography exclusion [14] , kinetic colorimetry [15] or else intracavity laser spectroscopic [16] . However, despite a higher cost compared to other techniques (due to the consumables including gas, input equipment maintenance, etc.), inductively coupled plasma mass spectrometry technique (ICP-MS) has become predominant in ultra-trace and multi-element analysis thanks to its attractive features like high selectivity, sensitivity, accuracy, low detection limits and its ability to discriminate the isotopes of the same element according to their mass [17] . Even so, the lower the half-life (T1/2), the higher its specific activity should be. Therefore, the use of ICP-MS in radiotoxicology is mainly limited to stable and long-lived radionuclides (T1/2 > 10 years), such as 127I or 129I for iodine. For short-lived radionuclides (for instance, 123I, 125I or 131I), radiometric methods will be preferred [18] .
Iodine measurement by ICP-MS in various matrices (such as foodstuffs or biological fluids) has been already addressed in previous studies [19] [20] [21] [22] . For all these reasons, this analytical technique was naturally chosen for the measurement of iodine in the urinary matrix which was carried out by standard additions (referred as calibration 1) and external calibration methods (thereafter abbreviated as calibration 2). Moreover, recognizing that having access to facilities and authorizations required to manipulate radioactive iodine was challenging, we decided to manage our experiments with stable iodine (127I) only. This article aims to compare with two types of calibration for 127I determination, to report validation results and to present some data of urinary iodine in rodents (rats). This work will finally shed light on the most appropriate calibration method for urinary iodine determination not only in terms of accuracy but also for samples time preparation.
2. Materials and Methods
2.1. Instrumentation
Iodine determination was performed by ICP-MS using a quadrupole mass spectrometer “X Series II” (Thermo Fisher ScientificTM, France). From a ASX-520 autosampler (Cetac Technologies, USA), the sample is injected and transported by specific tubings through the sample introduction system composed by a standard peristaltic pump and a Meinhardt nebulizer fitting into a classic spray chamber. The sampler and skimmer cones used were in nickel. The skimmer cone was of type Xt more suited to loaded matrices. For each experiment, all instrument parameters controlled from PlasmaLab software version 2.6.3.340 (2007) were optimized daily using a tuning iodide solution (40 µg・L−1) to obtain the highest sensitivity and to minimize possible interference effects (oxide levels and doubly charged ions). To get a general picture, hereafter values of ICP-MS acquisition settings that we could have: forward Rf power (1403 W), pole bias (−6.00 V), hexapole bias (−1.70 V), cool gas flow (13.02 L・min−1), auxiliary gas flow (0.80 L・min−1) and nebulizer flow (0.76 L・min−1).
Iodine was determined at m/z = 127 and tellurium (Te), as internal standard, at m/z = 125 (125Te) in the pulse counting mode of data acquisition.
2.2. Chemical and Standard Solutions
Ultrapure water used throughout all experiments was obtained from a Milli-Q® Synergy 185 water purification system (Millipore, Saint-Quentin-en-Yvelines, France) with a resistivity about 18 MΩ. cm. Employed reagents were obtained from different suppliers and selected after careful checking of their low 127I concentration. All necessary stock solutions were prepared the day of the experiment.
1) In this present work, 25% ammonia solution Suprapur® (MERCK MILLI- PORE, Fontenay-sous-Bois, France) and 20% Normapur® (PROLABO, Fontenay-sous-Bois, France), of analytical-grade were used. To dilute the samples, we prepared 2% ammonia hydroxide solution (2% NH4OH) by dissolving appropriate volumes of 25% ammonia solution Suprapur® in ultrapure water. In contrast, during measurements, the rinse solution used for ICP-MS between samples consisted of 5% ammonia hydroxide solution prepared from 20% ammonia solution Normapur®.
2) Regarding iodine oxidation state in solution which is a critical issue for iodine determination, to avoid iodide (I−) oxidation and then to stabilize these ions [23] , we chose to add a strong reducing agent, sodium thiosulfate pentahydrate Suprapur® of analytical-grade (MERCK MILLIPORE, Fontenay-sous-Bois, France). A sodium thiosulfate stock solution was prepared at a concentration of 184 mg・L−1 in 2% ammonia solution.
3) To correct any signal intensity variations, 125Te was selected as internal standard because of its close physicochemical properties to iodine (particularly atomic mass and first ionization energy). ICP standard of Te (1000 mg・L−1) in 2% - 3% nitric acid (MERCK MILLIPORE, Fontenay-sous-Bois, France) was purchased and this standard was diluted in 2% ammonia solution to reach a 125Te concentration of 1.25 mg・L−1 (or 17.68 mg・L−1 in total Te).
4) For the calibration range of our two methods, from iodide certified standard solution in aqueous solution commercially available (1000 mg・L−1) (VWR, Fontenay-sous-Bois, France) stored at 2˚C - 8˚C, the iodide calibration solution at 100 µg・L−1 was prepared by performing two successive dilutions. For both methods, the calibration range included six points between 0.4 and 8 µg・L−1 (0.4 ± 0.010, 1.0 ± 0.019, 2.0 ± 0.110, 4.0 ± 0.146, 6.0 ± 0.175, 8.0 ± 0.186).
2.3. Reference Materials
The validation of the methods has been carried out with certified reference materials (CRM, SeronormTM Trace Elements Urine, ref. 210305 and 210705) obtained from Ingen, France. As no CRM rat urine was unfortunately commercially available to our knowledge, we used human urine CRM to perform the validation. These CRMs, stored at 2˚C - 8˚C, are lyophilized human-based control materials produced from voluntary donors. As recommended, a volume of 5 ml of ultrapure water was added in the urine in order to reconstitute them. To confirm the applicability of our analytical methods, two levels of I− certified concentration in urine are commercially available, 84 ± 6 µg・L−1 and 304 ± 22 µg・L−1. Furthermore, validation process was also performed at an iodide concentration of 42 ± 2.95 µg・L−1, from the CRM at 84 µg・L−1 after a 2-fold dilution in ultrapure water. Before ICP-MS measurement, total dilution factors of 63 (for CRMs at 42 and 84 µg・L−1) and 125 (for CRM at 304 µg・L−1) were applied respectively.
2.4. Iodine Analysis Procedure
2.4.1. Calibration 1 (Standard Addition)
For this method, the first step involved the preparation of a pool composed of sodium thiosulfate (184 mg・L−1), 125Te and urine CRM. Then, the standard additions were prepared. For this, 1.2 mL of the pool was added in a 15-mL light sensitive centrifuge tube (VWR, Fontenay-sous-Bois, France) to the appropriate amounts of iodide solution to obtain 10 mL calibration solutions of concentrations between 0.4 and 8 µg・L−1, the tubes were finally filled up to 10 mL with 2% ammonia solution. Table 1 summarizes the standard additions preparation.
2.4.2. Calibration 2 (External Calibration)
For calibration range (Table 2), 100 mL volumetric flasks were used (VITLAB GmbH, Grossostheim, Germany) in which appropriate volumes of iodine standard solution (100 µg・L−1) were added to the pool to get a calibration range from 0 to 8 µg・L−1. The calibration solutions contained also 125Te and sodium thiosulfate solution (18.4 mg・L−1).
The sodium thiosulfate solution (18.4 mg・L−1) was prepared in the same conditions as calibration 1. Indeed, from the beginning to the end of the sample preparation, sodium thiosulfate solution underwent a 10-fold dilution.
For sample preparation, CRMs were diluted in 15-mL light sensitive centri-
![]()
Table 1. Standard additions preparation.
![]()
Table 2. External calibration standard solutions preparation.
fuge tube by using the adequate dilution factor (63 or 125 depending on the iodine concentration) with 125Te, and sodium thiosulfate solution (18.4 mg・L−1) in the same proportions as for the calibration range.
Both established analytical methods required 300 seconds for sample analysis and 300 seconds for rinsing the equipment; such precautions were taken to maintain the lowest possible iodine signal (around 400 cps). For statistical purposes, each sample was prepared three times, and therefore in total thirty replicates of each sample were obtained. Finally, for calibration 2, the reagent blank signal was subtracted from each sample signal.
2.5. Methods Validation Process
The validation is an essential process towards the implementation of a new analytical method. This represents a quality assurance pledge and thus allows the laboratory to assess data reliability for further interpretation. In the present work, the validation consisted of evaluating the relevant performance criteria. To this end, the laboratory relied on several regulatory standards [24] , offering methodological principles and recommendations. The lab’s challenges were as follows: 1) to investigate the calibration function (linearity), 2) to study the accuracy of the method, 3) to assess the limit of detection (LOD) and the limit of quantification (LOQ) and to check the previously measured LOQ, 4) to determine the specificity by characterizing the yields and 5) to avoid any cross-contamination. All the previously mentioned criteria studied for both proposed methods have required several assays and then were evaluated using statistical tests as described below.
2.5.1. Linearity
For the linearity study, six and seven sets of experiments for calibration 2 and calibration 1 methods respectively, were performed on six calibration standard solutions (0.4, 1, 2, 4, 6 and 8 µg・L−1) and each experiment was repeated three times. To validate the linearity range, a statistical fit test (Fisher) was applied. It compared the observed model error and the observed experimental error using the analysis of variance, considering a normal distribution of the data.
2.5.2. The accuracy of the Method
The measurement accuracy is the closeness of agreement between the measured quantity values to that quantity’s true value. The study of accuracy implies the evaluation of intermediate precision and measurement trueness expressed in terms of bias with respect to a reference value. The intermediate precision which represents the intra-laboratory reproducibility consists of analyzing a same sample under different conditions by varying for example at least one of the factors such as the operator or the time. Finally, the accuracy interpretation was based on the Fisher F test conclusion that addresses also the experimental condition changes.
To assess the accuracy of the method, the validation plan for calibration 2 consisted of six sets of experiments on the six calibration standards (0.4, 1, 2, 4, 6 and 8 µg・L−1) and three experiments per set per level, resulting to a total of one hundred and eight values. For calibration 1, seven sets of experiments were taken into account with six levels of concentration and three experiments per set per level resulting to a total of one hundred and twenty six values.
2.5.3. Limit of Detection and Limit of Quantification
LOD is the lowest concentration of the analyte to be detected but not quantified, and LOQ is the lowest concentration of the analyte to be quantified, under defined experimental conditions. They were determined respectively as three times and ten times the standard deviation of the concentration in ten reagent blank aliquots (with 30 replicates per sample). Then, the results were multiplied by the appropriate dilution factor to obtain LOD and LOQ in the sample matrix.
Once LOQ defined, the objective was to verify the accuracy of the measured LOQ within an acceptable maximum deviation of 60% by checking Inequalities (1) and (2):
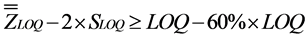 (1)
(1)
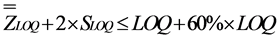 (2)
(2)
The parameters “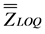 ” and “
” and “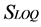 ” correspond to the overall mean and the intermediate precision standard deviation, respectively. The value of 60% was defined by convention [24] .
” correspond to the overall mean and the intermediate precision standard deviation, respectively. The value of 60% was defined by convention [24] .
To test the LOQ, the reagent blanks were spiked with iodine at a concentration equal to the LOQ defined above. Five sets were considered in intermediate precision conditions (for calibration 1, four experiments were performed) with four repetitions per set.
2.5.4. Specificity
The specificity of a method is its ability to measure only the analyte of interest. The analysis of this criterion allows knowing whether the measured response was disturbed by physico-chemical species other than the analyte of interest. Indeed, these constituents may be an important source of systematic errors.
To verify the absence of interfering substances, seven and five sets of experiments (each experiment was repeated three times) for calibration 1 and 2, respectively were considered. According to the regulatory standards [24] , it is advisable to choose additions representing 20% and 80% of the highest calibration concentration. For calibration 2, iodine considered concentrations were 1.6 and 6.4 µg・L−1. For calibration 1, due to technical problems, three additions were envisaged, 0.67, 1.33 and 2.43 µg・L−1 corresponding to about 8%, 17% and 30% of the highest calibration concentration.
Then, for each experiment, the yield (or ratio between the observed and expected concentration of the addition) was calculated as well as the yield mean. Finally, the calculated recovery rates (referring to the overall yields, for a given addition) were considered acceptable within 90% - 110% of expected spike value.
2.5.5. Study of Possible Cross-Contamination
To identify a possible cross-contamination, for both calibration methods, an experiment was conducted by analyzing three samples at 8 µg・L−1 and then three samples at 0.4 µg・L−1. Afterwards, a contamination value was calculated accor- ding to the following Equation (3):
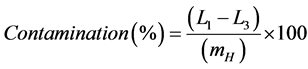 (3)
(3)
with L1, L3 and mH referring to the concentration of the first measured sample at 0.4 µg・L−1, the concentration of the third measured sample at 0.4 µg・L−1 and the mean value of the highest calibration standard, respectively.
Finally, the contamination value was compared to a performance criteria set at 5%.
2.5.6. Urinary Iodine Estimation in Rats
Animal experiments were performed in 3-month-old male Wistar rats purcha- sed from Charles River Laboratories (France) and were handled according to the French Legislation and the European Directives regarding the care and use of laboratory animals. Experimental protocols were validated by the IRSN’s Ethics Committee. Rats were caged in metabolic cages for a 2-day acclimation period prior the experiment. They had free access to drinking water and food (with granules A04 having iodine content of 0.3 mg・kg−1 from SAFE, Augy, France) before and throughout the experimental period. During experiment six rats with normal iodized diet were gavaged once with 1 mL of injection water at pH 7.4 (control rats) and six rats with normal iodized diet were gavaged once with 1 mL of 0.35 g・L−1 potassium iodide (French Army Central Pharmacy, Orléans, Fra- nce) (treated rats). The 24-hour rat urines were collected at different times: for half of the control and treated rats, urine samples were collected 24 hours after gavage. For the other rats, urine collection was performed 48 hours after gavage. All urine samples were frozen at −18˚C. Then, urine samples collected a few months earlier were analyzed in triplicate using both methods, after dilution in 2% ammonia solution (400-fold dilution for control rats with both methods and relating to treated rats; for urines samples collected 24 hours and 48 hours after gavage, factors dilution applied were 7000 and 700, respectively for both methods).
2.6. Statistical Analysis
The experimental data were analyzed using GraphPad software version 5.0.
Once all experiments were carried out, prior the evaluation of each criteria, the distribution of the measured data was examined by using Shapiro-Wilk W test. A Gaussian distribution was observed for all data at each concentration level. Relating to Wilcoxon W test employed for comparison of both methods on rat urine samples, it is considered that no statistically significant difference is observed between the results when p > 0.05.
3. Results and Discussion
3.1. Memory Effects
Because of its physico-chemical characteristics, iodine determination requires special precautions in order to prevent any losses and memory effects particularly on the instrument. The first issue concerned the material used for sample preparation which constituted a critical step. To avoid any sample pollution, the utilization of single use material was proved to be necessary.
The second question raised was about the sample dilution medium. It is well known that in acid media, iodine is less stable [25] . Moreover, with iodate (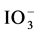 ) or I− form, highly volatile molecular iodine (I2) is generated. To overcome this problem, it is recommended to dilute the samples in alkaline solution to ensure the iodine chemical stability [26] . For this purpose, three basic solutions were investigated: sodium hydroxide (NaOH), tetramethylammonium hydroxide (TMAH) and NH4OH. The first was promptly abandoned because sodium ions cause a decrease of plasma temperature affecting iodine ionization. Sample dilution in NH4OH was chosen as on one hand iodine background was 2.5 times lower than the one with TMAH and as on the other hand, after the run of a highly contaminated sample (approximately 40 µg・L−1) the time requested to find a satisfactory background was faster with NH4OH.
) or I− form, highly volatile molecular iodine (I2) is generated. To overcome this problem, it is recommended to dilute the samples in alkaline solution to ensure the iodine chemical stability [26] . For this purpose, three basic solutions were investigated: sodium hydroxide (NaOH), tetramethylammonium hydroxide (TMAH) and NH4OH. The first was promptly abandoned because sodium ions cause a decrease of plasma temperature affecting iodine ionization. Sample dilution in NH4OH was chosen as on one hand iodine background was 2.5 times lower than the one with TMAH and as on the other hand, after the run of a highly contaminated sample (approximately 40 µg・L−1) the time requested to find a satisfactory background was faster with NH4OH.
Finally, the cost of TMAH is not negligible representing an additional point to consider.
As we can notice, everything is done to preserve the stability of iodine during the analysis. Nevertheless, in order to verify the absence of any pollution in ICP-MS experiments, we regularly place a reagent blank (2% NH4OH). In this way, we assure to return to an iodine background (approximately 400 cps).
3.2. Internal Standard
It is well known that urine constitutes a complex matrix due to the presence of organic and inorganic substances. That is why, the choice of the internal standard is a key element because its spike in the sample is intended to correct any signal drift and matrix effects. According to the literature, two standards are regularly employed in iodine measurement: iridium 193 (193Ir) and tellurium 125 [27] . A first set of experiments with urine samples spiked with these both internal standards shows that 193Ir was not stable over time under our experimental conditions. In addition, its mass and its potential first ionization do not make it as a good candidate. Regarding our two methods, we used the results with and without 125Te correction. It turned out that for the standard addition method, 125Te allows to efficiently correct the instrumental drift and thus to be closer to the expected values. For this calibration method (1), 125Te correction has been thus applied to all samples signals.
Nevertheless, for calibration 2 (external calibration), the sample comparison of the signals obtained with and without 125Te correction showed that the 125Te correction was not appropriate. Along ICP-MS measurements, the signal at mass 125 fluctuated too widely in the standard solutions medium (sodium thiosulfate in 2% NH4OH) compared to our matrix of interest (urine), signal drift and matrix effect could not be offset. In most cases, the relative biases calculated by taking into account 125Te correction were farther the target value. Consequently, for calibration 2, we refrained to use 125Te correction.
3.3. Validation Process
3.3.1. Linearity
Measured iodine concentrations are compared to expected ones in Table 3 and Table 4. A very good agreement is observed since the ratio for each calibration point is close to 1. Concerning calibration 2, a slightly greater dispersion is noted at the highest concentrations and explains a lower coefficient of correlation value (0.9993) but still remains acceptable.
To verify the calibration curve linearity, the Fisher F test was implemented to test the hypothesis of non-validity of the linear range. The observed F value was compared with a F value from Fisher’s table with a α error risk of 1% and with the degrees of freedom p and p(n − 1) (p representing the number of standards and n the number of replicate per experiment).
The observed F values were 0.81 (with Ftable = 3.35) and 0.45 (with Ftable = 3.47) for calibration 1 and calibration 2 respectively. For both, the calculated F value was less than the F table value at the risk of 1%. Therefore, the model error was
![]()
Table 3. Calibration 1: ratios between expected values and measured values.
![]()
Table 4. Calibration 2: ratios between expected values and measured values.
negligible compared to the observed experimental error. The assumption of non-validity of linearity range was denied. Thus, the calibration curves are considered linear in the concentration range investigated.
3.3.2. Accuracy
Before assessing accuracy parameters, it is recommended to apply the Cochran C and the Grubbs G statistical tests to identify the potential presence of aberrant or suspicious data. The Cochran C test examines the homogeneity of variances while the Grubbs G test explores the homogeneity of means. These tests were employed for each standard of both methods. The results have shown that the calculated values were all below the critical values at risk 5% and 1%, meaning that no doubtful value was detected. The next step consisted of determining the precision by focusing on the coefficients of variation of repeatability (CVr) and intermediate precision (CVR) and the evaluation of the trueness through the bias (corresponding to the systematic error estimation).
The most relevant results are reported in Table 5 and Table 6.
First of all, as shown in Table 5 and Table 6, CVr and CVR are very satisfactory with values below 5% (excepted for one value for calibration 2) implying a closeness of the measurements around the mean and so reflecting methods of very good precision. Then, relating to the trueness, as noticed, according to Equation (4), relative biases calculated varied from −0.56% to 0.44% for calibration 1 and ranged from −1.37% to 0.58% for calibration 2.
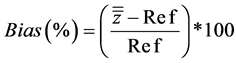 (4)
(4)
with Ref being the target value and 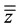 the mean of the means.
the mean of the means.
Bias interpretation is based on normalized deviation (ND) which is described by the Equation (5) below.
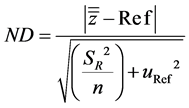 (5)
(5)
with SR2 referring to the intermediate precision variance, n corresponding to the number of experiments and uref relating to the uncertainty of the target value.
As illustrated in Table 5 and Table 6, all biases are widely less than 2% suggesting that biases are considered statistically negligible. However, the accuracy study also requires verifying Inequalities (6) and (7), involving the upper/lower acceptance and tolerance limits.
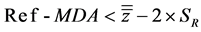 (6)
(6)
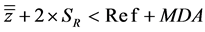 (7)
(7)
with MDA meaning Maximum Deviance Acceptable.
The terms “Ref + MDA” and “Ref ? MDA” refer to the upper and lower acceptance limits, respectively whereas “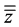 + 2 × SR” and “
+ 2 × SR” and “ −2 × SR” relate to the upper and lower tolerance limits, respectively.
−2 × SR” relate to the upper and lower tolerance limits, respectively.
These previously mentioned inequalities were calculated for each level of standard and we can observe that the tolerance intervals are comprised within the acceptability limits. In all cases, the accuracy was confirmed.
Nevertheless, to strengthen these conclusions, it is necessary to perform the Fisher F test which judges the influence of modified conditions (operator, time). The results obtained are tabulated in Table 5 and Table 6 and are all below F value from Fisher’s table at risk 5% (2.848 and 3.106 for calibration 1 and 2, respectively), confirming that conditions changes are negligible and do not affect
![]()
Table 5. Accuracy assessment for calibration 1.
![]()
Table 6. Accuracy assessment for calibration 2.
the results. Finally, for each calibration point, the expanded relative uncertainty was evaluated. For calibration 1, the results fluctuate between 7% and 2% going to the lowest to the higher iodine concentration whereas for calibration 2, the uncertainties are larger with values ranging from 11% to 5%. These expanded relative uncertainties are quite satisfactory.
3.3.3. LOD and LOQ Assessment
LOD and LOQ experiments were conducted on ten reagent blank samples (containing sodium thiosulfate, 2% NH4OH and 125Te). Once analyzed, through the calibration function, iodine concentrations were determined as well as the standard deviation on all measurements. Then, LOD and LOQ in urine were evaluated taking into account the dilution factor used (63-fold) for the samples. LOD was estimated to 0.39 µg・L−1 for calibration 1 and 0.35 µg・L−1 for calibration 2.
About LOQ, the values found were 1.30 µg・L−1 and 1.18 µg・L−1 for calibration 1 and 2, respectively.
Literature reported on iodine LOD in urine by ICP-MS are quite various. Among them, Allain et al. determined a LOD (calculated as twice the standard deviation of the background signal) in urine of 1.6 µg・L−1 with a 10-fold dilution in acid media using external calibration method [19] . Other authors have carried out measurements on twenty urine samples with a 50-fold dilution factor in alkaline media using external calibration and matrix-matched calibration solutions [28] . The LOD estimated was 1 µg・L−1. Finally, according to another recent publication, a LOD was determined at 4 µg・L−1 on blank samples in acid media [29] . Compared to these works, our two proposed methods exhibited very satisfactory LOD values.
To complete the validation process, the verification of the LOQ previously measured (0.02 µg・L−1 considering the sample dilution factor) was performed. For calibration 1, as the previously mentioned value could not be verified, we chose the value 0.2 µg・L−1 while remaining below the lower calibration standard solution (0.4 µg・L−1).
The results for the two methods are presented in Table 7. For each calibration methods, the two inequalities were verified.
3.3.4. Selectivity
Before analyzing the results, as for accuracy, the Cochran C test and the Grubbs G test were employed at each addition and for both methods. All data followed a Gaussian distribution. Therefore, selectivity analysis could be performed. The results are reported in Table 8.
For calibration 1, the objective was to reach the values 0.67, 1.33 and 2.43 µg・L−1. The achieved recovery rates were respectively 98.3%, 95.4% and 93.1%. The results are considered quite sufficient since they are within 90% - 110% of expected value.
![]()
Table 7. Results of the verification of LOQ calculated before for calibration 1 and 2.
![]()
Table 8. Specificity results for calibration 1 and 2.
For calibration 2, the values 1.6 µg・L−1 and 6.4 µg・L−1 were investigated and the estimated average yields were 107.1% and 108.4%. These results are more and less 10% from the value targeted which is acceptable.
3.3.5. Cross-Contamination Study
Possible contamination may occur and affect the samples during the analysis. An evaluation of the inter-sample contamination is therefore required to ensure the reliability of the results. The results were 0.03% and 0.15% for calibration 1 and 2, respectively. Thus, as the performance criterion is set at 5%, the results are more than suitable.
3.3.6. Urinary Iodine Estimation in Rats
The concentration of urinary iodine of twelve rat urine samples (six control rats and six treated rats) was determined by ICP-MS using both calibration methods previously validated.
The measured values mean results are shown in Table 9.
U1 refers to overall uncertainty for calibration 1 (u1, u2 and u3 correspond to uncertainties for the three replicates) and U2 refers to overall uncertainty for calibration 2 (u1, u2 and u3 correspond to uncertainties for the three replicates).
A high discrepancy is observed in iodine concentration in urines from treated rats. For rats n˚100, 120 and 140, 24-hour urine samples were collected 24 hours after gavage which explains the high concentrations of iodine. Indeed, after absorption, iodine is rapidly excreted [30] whereas, for the rats n˚102, 122 and 142, 24-hour urine samples were collected 48 hours after gavage which justifies significantly lower iodine concentrations.
To compare the results of both calibration methods, the non-parametric Wilcoxon W test and the Z-score calculation were used. For each sample, as noticed in Table 9, Wilcoxon W test concluded to no significantly difference between the results. Consequently, according to this test, both calibration methods are thus comparable.
Relating to the other way to investigate the results, namely Z-score, the interpretation of this parameter is as follows. When the calculated Z-score is between −2 and 2, the result is considered as satisfactory whereas if a Z-score value is found between −3 and −2 or 2 and 3, the result is seen as doubtful. Considering the obtained values, the results are quite acceptable except for the treated rat n˚ 122 (with a Z-score of −2.23).
![]()
Table 9. Results of urinary iodine in rats by both calibration methods.
aDue to technical problem, mean is measured only on two replicates; bns means no difference statistically significant; c![]() with
with ![]() and
and![]() .
.
These two tests allow us to conclude that both calibration methods are comparable.
These analyses in control rats were the opportunity to estimate the urinary iodine in this animal species because as far as we know, the information on this biological parameter is of great rarity. According to our findings, urinary iodine concentration for 3-month-old male Wistar rat is comprised between 672.31 µg・L−1 and 1197.92 µg・L−1 and highlights the great intra-rat variability. Even though this information was widely available in the literature, there would be too much uncertainty related to the diet, the gender or the species to compare the data.
4. Conclusion
Taking into account the results of each criterion, we can conclude that both calibration methods for urinary iodine determination by ICP-MS are validated as analyzed performances fully complying with all the requirements we had previously imposed, with maybe greater satisfaction for calibration 1 (especially considering its very good linearity, excellent accuracy with quite low expanded standard uncertainties). Given the validation results, both calibration methods can be used. Nevertheless, if focus is given on practical or logistical issues, external calibration method should be preferred because of an overall analysis time significantly shortened and therefore a quicker delivery of the results. To these advantages, we can also add the lowest cost for lab materials. Otherwise, the recent acquisition of an ICP-MS “iCAP-Q” (Thermo Fisher ScientificTM, France) enabled us to considerably optimize the analysis time (factor 9 between ICP-MS “X series II” and “iCAP-Q” in external calibration). In fact, the device is equi- pped with a FAST system which starts rinsing even though the sample is being introduced into the ICP-MS. Finally, when applied to iodine measurement in rat urine samples, both calibration methods are in good agreement. Thus, we can conclude that both procedures have been validated successfully ensuring the reliabilities of future results for rodent’s experiments.
Acknowledgements
This study has been partly supported by the French National “Investment for the future” funding programme. The authors are indebted to David Suhard, Guill- aume Phan and Rym Chioukh for the supply of rat urine samples.