Journal of Biophysical Chemistry
Vol.2 No.2(2011), Article ID:5137,10 pages DOI:10.4236/jbpc.2011.22013
Flow cytometric investigation on degradation of ma-cro-DNA by common laboratory manipulations
1Center for Bioanalysis, Korea Research Institute of Standards and Science, Daejeon, Korea;
*Corresponding Author: srpark@kriss.re.kr
2Department of Chemistry, Chungnam National University, Daejeon, Korea.
Received 7 January 2011; revised 3 February 2011; accepted 11 January 2011.
Keywords: Physical Degradation of DNA;Laboratory Manipulations; Flow Cytometry;
Exhaustive Counting; Self-Protection Effect
ABSTRACT
The degree and characteristics of physical degradation of macro-DNA molecules by common laboratory manipulations are reported. With linearized lambda-phage viral DNA as the model DNA, fragmentation of macro-DNA by various indispensable laboratory manipulations were investigated using a high sensitivity flow cytometric setup. Investigated manipulations included pipetting, vortexing, rocking, freezethawing, ultrasonication and ultrafiltration. “Exhaustive counting” of the intact lambda DNA molecules following such manipulations enabled a quantitative assessment of the resulting fragmentation, which also revealed the type of degradation reflected in the fragmentation patterns. The use of high sensitivity flow cytometry was especially suited to investigate the degradation of dilute DNA solutions that may not be suitable for analysis using traditional methods. Notable findings of this study included: the boarderline-size of DNA chains in terms of susceptibility to shear stresses by such manipulations; discernable instability of nicked DNAs; shattering-fragmentation of DNAs by freezethawing or ultrasonication; effectiveness of some protection media; marked “self-protection effect” of concentrated DNA solutions. These findings support and refine our traditional knowledge on how to maintain the physical integrity of macro-DNA molecules against inevitable laboratory manipulations.
1. INTRODUCTION
Deoxyribonucleic acid (DNA) is an essential component of modern biology. Its size spans from several 10s of bp (base-pairs) of synthetic oligonucleotides to a few million bp of double-stranded DNA. Although DNA is known to have a robust chemical structure that can stably retain genetic information, large DNA molecules can easily be degraded by unattended physical experiments [1]. This could be an important concern when one has to maintain the integrity of DNA as a critical agent such as in the area of gene-therapeutics [2]. Multiple gene therapy strategies have recently began to use artificial chromosomes in the mega-base size range [3]. The handling of such large DNAs with minimal impairment of macromolecular integrity is of paramount importance in this field.
The fragility of long polymer chains to physical stresses was first demonstrated by Frenkel in 1944 [4]. Since then, knowledge concerning the theoretical aspects of shear-induced DNA breaks has steadily grown [1,5-14]. Various influencing parameters were methodically investigated and theoretically inferred. As reviewed by Lengsfeld and Anchordoquy [2], the most important parameters that influence shear-induced breaks of macro-DNA are strain rate, ionic strength, gas-liquid interface and turbulence, in addition to DNA molecular weight, size, and flexibility. Recently, concerns about DNA degradation have been raised in relation to the mass production of high quality plasmids for use in DNA therapeutic strategies [3,10,11,14]. Various bio-processing steps utilized in mass production such as lysis, solid removal, purification, and finishing would impart physical stresses that can induce undesirable degradation of target plasmids [3]. Although a substantial amount of knowledge has been accumulated regarding the physical degradation of DNA, it does not seem to be readily applicable to routine laboratory environments. The outcomes of methodical investigations were often presented with complicated mathematical equations that may not be intuitive to most staff employed in biology laboratories. In terms of the intensities of imparted physical stresses, the investigated steps relating to bio-processing may not be comparable to routine laboratory manipulations that include pipetting, vortexing, rocking, freeze-thawing, ultrasonication, and ultrafiltrations. Additionally, routine laboratory manipulations may involve the use of DNA solutions that are significantly more dilute than those solutions employed in a mass production environment. It is known that dilute DNA solutions are more prone to physical degradation due to a reduced “self-protection effect” [12,13].
In this work, the physical degradation of macro-DNA by the aforementioned routine laboratory manipulations was investigated in a quantitative manner. Linear lambda viral DNA (48.5 kbp) was used as the model macroDNA. Fragmentation of the model DNA in dilute preparations (e.g., 3 ng/mL or 0.1 pM) by application of each manipulation was quantitatively assessed. Various methods have been applied for the analysis of DNA fragmentation including gel electrophoresis [10,11,14], ultracentrifugation [1,5,7], electron microscopy [5], and even the use of a microplate-reader with fluorescence detection [11]. Relative to these methods, the high sensitivity flow cytometry [15] deployed in this work is not only rapid and convenient, but is also sensitive enough to analyze highly diluted DNA solutions. The flow cytometric system can detect a single DNA down to the size of 6 kbp in a flow stream. Intact lambda DNA and its fragmented products were to be counted together with their size information following each manipulation in an effort to reveal the degree and type of degradation. ‘Exhaustive counting’ that counts all meaningful DNA particles in a given volume was applied to accurately quantify the degree of fragmentation. This method was applicable to highly diluted DNA solutions (down to 3 ng/mL) so that the effect of reduced self-protection was clearly observable. The histograms of the applied flow cytometry presented DNA fragments in the size range between 6 and 50 kbp. The flow cytometric run time was typically 5 minutes. The common laboratory manipulations investigated were pipetting, vortexing, rocking, freeze-thawing, ultrasonication, and ultrafiltration.
2. MATERIALS AND METHODS
2.1. DNA Samples and Other Reagents
Linearized lambda phage DNA was purchased from Fermentas (#SD0011; Burlington, Canada). This DNA solution (0.3 mg/mL or 1 ´ 10–8 M) was used as the stock solution. The stock solution was diluted with 1 ´ TE buffer (10 mM TRIS, 1 mM EDTA, pH 8.0) down to 3 ng/mL (~1 ´ 10–13 M) to prepare test solutions. Unless otherwise indicated, the concentration of a test solution was 3 ng/mL. Test solutions were subject to various manipulations as described below, and small portions of untreated test solutions were stored as reference solutions for the counting measurement. For the counting experiment, the concentration of each test solution was adjusted to 3 ng/mL with 1 ´ TE buffer if the initial concentration was higher than this value. Then, 50 μL of the test solution (undiluted or following concentration adjustment) was further diluted into 1.5 mL of Tris buffer (pH 9.5) that included 10% DMSO and 5 μL 1 ´ SYBR® Gold dye (Invitrogen, Carlsbad, CA, USA).
2.2. Fragment Analysis by Flow Cytometry
The instrumental setup employed for the flow cytometry has been described elsewhere [15]. In brief, 50 μm ´ 50 μm square-type fused silica tubing (WWP050375; Polymicro Technologies, Phoenix, AZ, USA) was used as the flow channel. The total length was 1 m where the detection spot was established 60 cm away from the sample introduction-end of the tubing. The whole capillary channel was overfilled with a test solution. A thin DNA flow stream was generated by applying 0.03 MPa and –5 kV to the sample introduction-end, and each DNA particle passing the detection window was counted for 4 minutes. The DNA count reached a maximum typically in 2.5 minutes of counting. A laser induced fluorescence (LIF) detection system was setup to detect each dye-stained DNA particle when it passed the detection zone. The detection system was finely tuned to the narrowest possible peak for the intact lambda DNA count in a histogram. If necessary, histograms were changed to density plots for easy comparison of a data set from multiple experiments.
Each manipulation was applied to typically three aliquots of a test solution, and then the results of each aliquot were pooled to reflect possible variations in the intensity of physical stresses imparted by the given manipulation. Counting was repeated three times for a given aliquot, and the results were averaged. Before every measurement, a reference solution was counted to provide a reference data set. The averaged intact lambda DNA count for each measurement solution was compared to the reference data set to calculate the fraction of intact lambda DNA that withstood the stresses from the given manipulation. Then, the ‘degree of degradation’ by the given manipulation was defined as [1-the fraction of intact DNA]. In this calculation, the effect of possible variations in total DNA quantity due to inexact dilution was eliminated by normalizing the fraction of intact DNA based on the total DNA quantity.
2.3. Investigated Manipulations
2.3.1. Pipetting
A linearized lambda viral DNA solution (3 ng/mL in 1 ´ TE buffer) was repeatedly pipetted a number of times as described in the text using Gilson (Midleton, WI) pipettes with capacities of 0.1 or 1 mL to draw 0.07 or 0.7 mL, respectively. Additionally, an Eppendorf Multipette plus pipette (model #4337806; Hamburg, Germany) was also employed in drawing 0.7 mL of the DNA test solution. A decade-cascade-dilution series of the original 0.3 mg/mL (~1 ´ 10–8 M) solution down to 3 ng/mL (~1 ´ 10–13 M) was also subject to 10 repeats of 0.7 mL/1 mL pipetting with the Gilson pipette to investigate the self-protection effect in this concentration range. Gentle (soft) pipetting was done by slow drawing and dispensing (for 5 seconds for each operation) with a pause for 5 seconds in between them. Inattentive (rough) pipetting was performed in a rapid manner (for 0.5 seconds for each operation) without giving a pause in between them.
2.3.2. Vortexing
1 mL of the dilute lambda viral DNA solution above (3 ng/mL in 1 ´ TE buffer) contained in a 2 mL glass vial (#5182-0716; Agilent, Santa Clara, CA) was vortexed using a typical benchtop vortexer (Vortex Genie 2; Scientific Industries, Bohemia, NY). Vortexing was performed for 10, 30, 60 or 180 seconds. Various power levels were tested as described in the text.
2.3.3. Rocking
The lambda viral DNA solution above (3 ng/mL in 1 ´ TE buffer) was placed in a benchtop rocker (#SLRM-2M; Seoulin Bio, Seoul, Korea) and rocked for 10, 30, 60, or 120 seconds at 10, 30, or 60 rpm (in a simple halfway-rotation mode). The sample volume and the container was the same as in the vortexing experiment.
2.3.4. Freeze-Thawing
The same lambda viral DNA solution above (3 ng/mL in 1 ´ TE buffer) was repeatedly frozen and thawed between –20˚C and room temperature for 1, 3, 6, 10, 15 and 20 times. Additionally, the decade dilution series as in the pipetting experiment was repeatedly frozen and thawed in the same manner for 10 or 20 times to investigate the self-protection effect.
2.3.5. Ultrasonication
The same lambda viral DNA solution above (3 ng/mL in 1 ´ TE buffer) was ultrasonicated for 10, 30, 50, 120, 240, or 360 seconds using a bench-top ultrasonicator (#UC-10; Jeio Tech, Daejeon, Korea). Two power levels of ‘low’ and ‘high’ were applied. The same lambda viral DNA solution but containing 10% DMSO was also applied to the ultrasonication test.
2.3.6. Ultrafiltration
0.5 mL of the original preparation of the linearized lambda viral DNA solution (0.3 mg/mL in 1 ´ TE) was placed in a Microcon YM-3 or YM-30 filter (Millipore, Billerica, MA) and centrifuged for 100 minutes at 13000 rpm using a bench-top micro-centrifuge (#D-37520, Thermo Scientific, Waltham, MA). The DNA on the filter membrane was released by soaking in 1 ´ TE buffer for one day. The recovered solution was diluted and assessed by flow cytometry.
3. RESULTS AND DISCUSSION
3.1. Flow Cytometric Assessment of DNA Degradation (Fragmentation)
A typical histogram for a reference solution of intact lambda phage DNA (48.5 kbp) is shown in Figure 1(a). Typically, about 95% of the total observed DNA represented intact lambda DNA. The linear relationship be-

Figure 1. Flow cytometric measurement of DNA fragments.(a) Typical histogram of a reference DNA sample (linearized lambda phage DNA prior to application of physical stresses, 0.1 ng/mL lambda DNA in 1.5 μL); (b) Typical histogram of X-bal-digested lambda DNA; (c) Typical histogram of Hind III-digested lambda DNA; (d) Linear relationship between expected DNA fragment size and fluorescence intensity (the x-axis scale of a histogram) extracted from the results above.
tween DNA size and fluorescence intensity (the x-axis of the histogram) was confirmed from the histograms of the X-baland Hind III-digested lambda DNA products (Figures 1(b) and (c), respectively). The expected DNA fragment following X-bal digestion is 24.5 kbp, whilethose for Hind III digestion are 25.4, 23.1, 9.42, and 6.55 kbp. As shown in Figure 1(d), the x scale was linearly correlated with DNA size (R = 0.995). DNA fragments down to 6 kbp could be identified on the histogram. ‘Exhaustive counting’ was applied, and counting results showed good reproducibility with a RSD below 2%. ‘Exhaustive counting’ refers to counting all DNA particles in the sample volume, which results in high reproducibility as the counting results are unaffected by variations in the operation conditions of the measurement system.
3.2. Manipulation-induced Degradation
3.2.1. Shear Degradation by Pipetting, Vortexing, Rocking, or Ultrafiltration
Among tested manipulations, pipetting, vortexing, rocking, or ultrafiltartion would possibly result in DNA degradation from shear stress as it arises from violent flow of a viscous medium. Shear stress involves the whole mass of a DNA chain, which would be concentrated on the mass center of the DNA chain [1,4-6]. The initial outcome would be the breakage DNA chain to two approximately half-sized DNA fragments, which was experimentally observed in this work. The results of this experiment confirmed that inattentive operations of such manipulations could lead to a substantial compromise of the integrity of macro-DNAs such as lambda phage DNA (48.5 kbp).
Inattentive pipetting led to dramatic degradation of lambda phage DNA, especially when the pipetted volume was large (0.7 mL using a 1 mL-pipette). The degree of fragmentation apparently depended on the gentleness of the drawing-dispensing action of the pipette. For example, when 0.7 mL of a dilute DNA solution (3 ng/mL) was carelessly drawn and dispensed using an Eppendorf Multipette plus pipette (model #4337806), more than 70% of the intact lambda DNA became fragmented (Figure 2(a)). This particular pipette showed extraordinary stiffness in its moving mechanism, which led to rather rough pipetting actions. Although attentive, gentler operation resulted in reduced fragmentation, more than 50% of the lambda DNA became fragmented by a single pipetting action. Even when utilizing other types of pipettes with smooth moving mechanisms, inattentive pipetting led to substantial fragmentation that was correlated to the number of repeats of pipetting (Figures 2(b) and (c)), and almost complete fragmentation (up to 90% degradation) had occurred. Fragmented

Figure 2. Degradation of lambda DNA by pipetting. (a) Density plots of a lambda DNA solution prior to and following pipetting using a Eppendorf Multipette plus pipette (model #4337806) featuring an extraordinarily stiff drawing/dispensing mechanism; (b) Progress of degradation along the repeats of inattentive pipetting of a 0.7 mL test solution using a 1 mL pipette (Pipetman P-1000 from Gilson). Numbers at the right side indicate the number of pipetting applied to a dilute DNA solution (3 ng/mL); (c) Plot of the results in panel B; (d) Self-protection effect against inattentive pipetting. Lambda DNA solutions of various concentrations were subjected to 10 repeats of inattentive pipetting (0.7 mL with a 1 mL pipette).
DNAs showed a pattern comprising a broad peak centered at the half of the size of the intact lambda DNA (Figures 2(a) and (b)) as mentioned above. In the case of careful pipetting that would allow virtually turbulence-free drawing and dispensing with a sufficiently
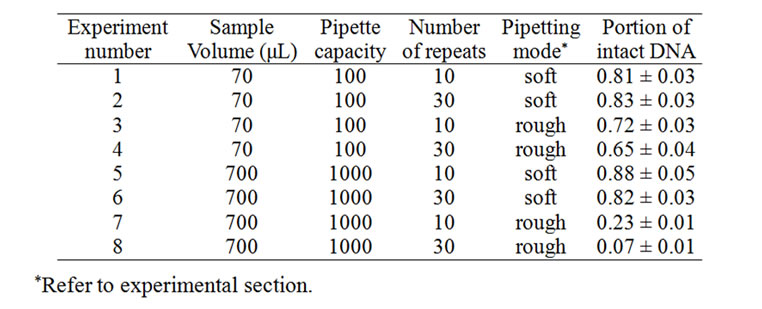
Table 1. Degradation of 3 ng/mL linearized lambda phage DNA in 1 ´ TE buffer by repeated pipetting. The fraction of intact DNA surviving repeated pipetting is indicated with CV (n = 3).
long pause in between two actions, however, more than 80% of the lambda DNA remained intact even after 30 times of repeated pipetting (Table 1). Smaller pipetting volumes such as 70 μL using 100 μL pipettes showed similar survival of ~80% for careful repeats of 30 pipetting actions. In the case of inattentive pipetting of 70 μL, however, greater than 60% of the lambda DNA remained intact, whereas more than 90% became fragmented by inattentive pipetting of 700 μL.
Concentrated DNA solutions acquires protection against physical damage as is termed the “self-protection effect” [12,13]. The self-protection effect was prominent in this work. In contrast to the dilute lambda DNA solutions, concentrated solutions (down to 30 μg/mL) did not show any significant fragmentation by 30 repeats of casual pipetting using a 1 mL-pipette (Figure 2(d)). 30 μg/mL approximates 1 ´ 10–9 M of lambda DNA. The degradation of lambda DNA by pipetting could be completely or mostly obviated if concentrated as high as 30 μg/mL, but the protection effect rapidly diminished in the narrow range (0.3 - 30 μg/mL). Therefore, the concentration of the commercial lambda DNA package (0.3 mg/mL) seems appropriate. There has been some controversy concerning the self-protection effect. Dancis [7] as well as Adam and Zimm [8] did not observe the self-protection effect. Levy et al [14] also did not observe any concentration-dependent shear sensitivity of DNA, but observed the self-protection effect with shear with a liquid-gas interface. Therefore, the self-protection effect against shear damage may require a liquid-gas interface. Nevertheless, the self-protection effect was evident and dramatic for most manipulations tested in this work.
Vortexing for short time periods was not as detrimental as pipetting of a dilute lambda DNA solution (~3 ng/μL). Vortexing for 10 to 30 seconds at the maximum power using a typical benchtop vortexer still conserved about 80% of the intact lambda DNA. However, fragmentation ensued in a time-dependent manner with prolonged vortexing (Figure 3). Vortexing at the maximum power for 3 minutes led to about 70% fragmentation of


Figure 3. Vortexing-induced degradation of lambda DNA. (a) Comparison of densitograms of vortexed DNA (■) and reference DNA (○). A dilute lambda DNA solution (3 ng/mL) was vortexed at maximum power for 3 minutes; (b) Effect of vortexing power on lambda DNA degradation. X-scale indicates the vortexing power where the maximum power was delivered at the 10th level. A dilute lambda DNA solution (3 ng/mL) was vortexed at various powers for 3 minutes.
intact DNA and resulted in fragmentation patterns similar to those obtained for pipetting (Figure 3(a)). However, the degree of degradation gradually reduced to unobservable levels as the vortexing power level was lowered from the 10th (maximum) level to the 3rd level (Figure 3(b)). The threshold of physical stresses for DNA breakage seemed to coincide with the forces imparted on lambda DNA by such manipulations. Therefore, one could observe highly variable results depending on how these manipulations were performed with the lambda DNA. Wide variation in the degree of degradation was most prominent with pipetting (Table 1) and vortexing (Figure 3(b)). Such manipulations must be carefully carried out to minimize the physical stresses imparted to DNA. Operating on small volumes would be safer given the associated lower momentum of the sample solution.
It has to be understood that the shear stresses induced varies depending on the size of the DNA. For a given physical agitation, the imparted tensional force is proportional to the 1st to 2nd power of the molecular weight of a DNA particle [2]. For linear DNA chains, the exponent was found to be 1.2 by Bowman and Davidson [5]. This experiment indicates that physical agitation by the tested manipulations imparts forces on lambda DNA that is close to the chain strength of duplex DNA. Then, it is reasonable to suppose that DNA substantially smaller than lambda DNA would be quite resistant to degradation by these manipulations. As seen in Figures 2(a) and (b) and Figure 3(a), the population around half-sized lambda DNA did not diminish substantially even when most of the intact lambda DNA particles became fragmented, suggesting that stresses imparted on DNA particles of this size are not comparable to the DNA chain strength. We confirmed this expectation by observing no substantial degradation of X-bal-digested lambda DNA (24.5 kbp) following application of the aforementioned manipulations (data not shown). In mass production environment, smaller DNA (5 kbp plasmid) is found to be insensitive to shear-induced degradation [2,3]. On the other hand, DNA particles substantially larger than lambda DNA would be readily fragmented by these manipulations. The size of intact lambda DNA (linearized form) could be used as a borderline-size in considering the possible physical damage induced by these laboratory manipulations.
Machine-assisted rocking often employed for the homogenization of a DNA solution did not cause severe fragmentation of lambda DNA (Table 2). With speeds up to 60 rpm (1 rocking/s), fragmentation was limited to below 20%. Fragmentation was not augmented with increased speed or prolonged operation time under the given experimental conditions. Further fragmentation occurred only when substantially harsher manipulations were applied. Similar observations were made in gentle pipetting. As shown in Table 1, gentle (soft) pipetting did not degrade lambda DNA beyond 20% regardless of the pipetting volume or number of pipetting actions. It is likely that the fragmented DNA under mild agitations

Table 2. Degradation of 3 ng/mL linearized lambda phage DNA in 1 ´ TE buffer by machine-assisted rocking. The fraction of intact DNA surviving rocking agitation are indicated with CV (n = 3).

Figure 4. Fragmentation of lambda DNA by machine-assisted rocking. Distribution of fragmented DNA particles shows a less prominent peak of halfway broken lambda DNA. A dilute lambda DNA solution (0.3 mg/mL) was rocked at 30 rpm for 1 minute.
was in a substantially less stable state compared to the rest of the DNA that remained intact during the manipulations. The instability of the fragmented fraction would have resulted from nicking of the DNA chains. The 15% - 20% observed in this work is close to Bowman and Davidson’s estimation of the portion of lambda DNA with single-strand beaks (17% to 30%) [5]. This assumption was also supported by the histograms which showed less prominent peaks for about halfway broken lambda DNAs following application of mild manipulations (Figure 4). If nicking sites are rather randomly distributed, the length of the resulting fragments should display a wide distribution. Nevertheless, it is noteworthy that nicked DNA chains are prone to mild physical agitation and could be broken even by mild manipulations.
Ultrafiltration is often applied to purify macro-DNA molecules in an effort to remove small chemical contaminants. Although ultrafiltration involves high-momentum centrifugation, no significant degradation of lambda DNA was resulted with both M.W. 3 kD cut-off filters (YM-3) and 30 kD cut-off filters (YM-30) as shown in Figure 5(a). The recovered DNA from ultrafiltration did not show fragmented DNA particles that were larger than 10 kbp. As this experiment was performed with a concentrated DNA solution (0.3 mg/mL), this result could be attributed to the self-protection effect. Nevertheless, an ultrafiltration-cleanup step can be confidently employed without concern about fragmentation if the DNA solutions are sufficiently concentrated. Additionally, recovery from YM-30 filters was in general slightly higher (about 20%) than that obtained with YM-3 filters, which could be due to the ease of release of DNA from the filter membrane of larger pores. The filtrates from both filtrations did not show the presence of DNA particles larger than 6 kbp (Figure 5(b)).
3.2.2. Shattering Damage by Freeze-Thawing or Ultrasonication
Repeated freeze-thawing and ultrasonication were detrimental to dilute DNA solutions. In addition, fragmentation patterns (Figure 6(b)) were substantially different from those by shear-damaged DNA (Figure 2(b) and Figure 3(b)). Physical stresses from freeze-thawing as well as ultrasonication would be too highly localized to be associated with the whole mass of a macro-DNA chain. The intensities of such stresses would be insensitive to the relative position along the DNA chain. As a consequence, DNA chains would be shattered to many smaller fragments. The histograms of these manipulations show a continuous progression towards the disappearance of large DNA particles. Especially, prolonged ultrasonication did not leave any DNA particles of observable sizes (> 6 kbp with the given instrumentation). Unlike shear-damage, even much smaller DNAs would not be spared from such damages.
Initial cycles of repeated freeze-thawing led to severe


Figure 5. Flow cytometric analysis of ultrafiltrated lambda DNA solutions. (a) Densitograms of recovered DNA following ultrafiltration with M.W. cut-off filters of 3 kDa (●) and 30 kDa (□); (b) Densitograms of the pass-through solutions of the ultrafiltration experiment above. 0.3 mg/mL lambda DNA solution was centrifuged for 100 minutes at 13000 rpm, and then recovered after 1 day-soaking in 1 ´ TE.

Figure 6. Degradation of lambda DNA by repeated freeze-thawing. (a) Progressive degradation of DNA with increasing repeats of freeze-thawing; (b) Typical fragmentation patterns of repeatedly freeze-thawed lambda DNA solution: (□) reference solution; (▲) freeze-thawed 9 times; (◆) freezethawed 20 times. 3 ng/mL lambda DNA solution was used; (c) Self-protection effect against DNA degradation by repeated freeze-thawing. Freeze-thawing was applied 9 times (▽) or 20 times (▲) to lambda DNA solutions of various concentrations. 1 ´ 10–8 M (10 nM) is approximately 0.3 mg/mL. No additive was added to the solution matrix of 1 ´ TE buffer.
degradation of dilute DNA solutions. One cycle of freeze-thawing caused about 10% degradation of intact lambda DNA in TE buffer. As shown in Figure 6(a), the fragmentation of lambda DNA progressed in a repeat-dependent manner, reaching 75% degradation at 20-cycles of freeze-thawing. The self-protection effect was obvious in the tested concentration range. In contrast to pipetting (Figure 2(d)), the self-protection effect gradually diminished across the wide concentration range (3 ng/mL - 0.3 mg/mL) as seen in Figure 6(c). Repeated freeze-thawing that is unavoidable in many circumstances is quite detrimental, but its adverse effect can be reduced by storing DNA as concentrated as possible.
Ultrasonication is often applied to DNA solutions for the purpose of fragmenting DNA. Using a typical benchtop ultrasonicator, application of a ‘low’ level ultrasonication power for 10 or 30 s resulted in 30% and 70% DNA fragmentation, respectively where shattered
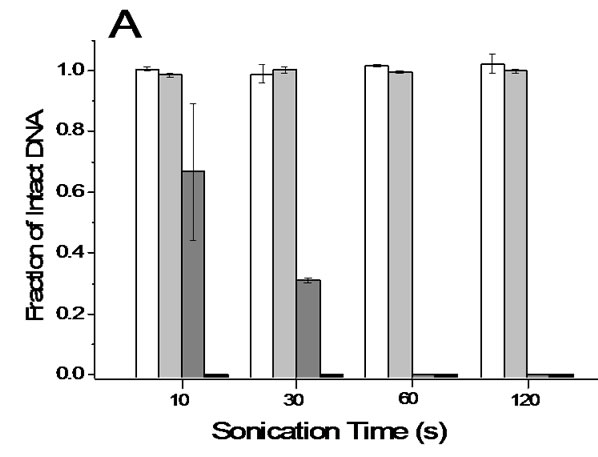

Figure 7. Degradation of lambda DNA by ultrasonication. (a) Protection effect of 10% DMSO. 3 ng/mL lambda DNA in 1 ´ TE buffer was markedly degraded by either weak (dark grey bar) or strong sonication (black bar) whereas the same DNA solutions containing 10% DMSO did not show any significant degradation following either weak (white bar) or strong sonication (light grey bar); (b) Shattering fragmentation of DNA by ultrasonication. Densitograms of sonicated DNA solution (●) and reference solution (□).
fragmentations were obvious (Figure 7). Application of a ‘high’ level ultrasonication power resulted in the complete absence of intact DNA, as was the case with prolonged ultrasonication at either power level. Therefore, ultrasoification has to be used only for purposeful fragmentation of DNA. It is noteworthy that DNA solutions containing 10% DMSO showed no observable fragmentation regardless of the power level and application times employed under the given experimental conditions. Intact lambda DNA particles in a DMSO solution were not noticeably fragmented even after ultrasonication for 10 minutes (data not shown). The protective effect of 10% (v-v) DMSO was also observed in the pipetting experiment. The fragmentation of nicked DNA by soft pipetting was completely avoided using a 10% (v-v) DMSO solution. If DMSO does not interfere with the procedures necessary for any given manipulation, the addition of DMSO would be a convenient way to reduce the physical degradation of macro-DNA. Other viscous media not tested here would provide a similar advantage. Concentrated NaCl (e.g. 150 mM) is known to provide protection effect against shear damage [16], that would be less prominent to linear DNA due to increased flexibility of the DNA chain by electro-neutralization [17]. Bowman and Davidson accordingly found insignificant protection effect of concentrated salt [5]. In this work, we observed marginal effect of 150 mM NaCl only for milder manipulations that disappeared with rough manipulations such as inattentive pipetting. Unlike shattering damage, DMSO neither effectively protected DNA against strong shear stresses. Therefore, violent manipulations need to be avoided even with such protection media.
3.3. Guidelines for Securing the Integrity of Macro-DNA
In summary of the observations and discussion above, the following guidelines are suggested for the handling of macro-DNA with reduced damage:
1) Use DNA solutions as concentrated as possible.
2) Keep in mind that ds DNA larger than 50 kbp is subject to degradation by unattended operation of common laboratory manipulations.
3) Avoid the handling of large sample volumes (including pipetting volumes).
4) Execute each operation as gently as possible to avoid sudden changes in motional direction of the sample liquid.
5) Be aware that nicked DNA is substantially more fragile.
6) Avoid freeze-thawing as much as possible. If unavoidable, prepare samples as concentrated as possible.
7) Consider the addition of an 'inert' protective medium (e.g. DMSO), but be aware their limited protection.
8) Ultrafiltration is a safe cleanup step when utilizing macro-DNAs.
The above guidelines may not be new to many experienced scientists. However, it would be better if the characteristics of DNA fragmentation are elucidated based on experimental data as our precautious measures could be optimized accordingly. In that sense, this experiment has a limitation in terms of tested DNA (only linearized lambda DNA). Differences in the size as well as the shape (ring, open chain or supercoil) of the DNA would substantially alter the observations. Therefore, further experiments will be carried out as soon as suitable materials and instrumentation are in hand.
4. CONCLUSIONS
Using a home-made flow cytometry system capable of counting individual DNA particles and providing information concerning DNA size, the fragmentation of linearized lambda phage DNA by common laboratory manipulations was examined in a quantitative manner. It was evident that large DNAs such as lambda phage DNA could easily be fragmented by the careless execution of several common laboratory manipulations, and especially when working with diluted DNA solutions. The degrees of damage associated with the physical strength of the execution of these manipulations were quantitatively presented, and provide helpful data in assessing the potential degradation of macro-DNA in a common laboratory environment. Although limited in the test DNA, the experimental findings in this work support and refine our traditional knowledge on how to minimize the physical damage of macro-DNA associated with common sample handling techniques.
5. ACKNOWLEDGEMENTS
This work was fully supported by the National Research Foundation of Korea as part of the research project, ‘New Technologies for Establishment of Measurement Standards for Biosciences’.
REFERENCES
- Davison, P.F. (1959) The effect of hydrodynamic shear on the deoxyribonucleic acid from T2 and T4 bacteriophages. Biochemistry, 45, 1560-1568.
- Lengsfeld, C.S. and Anchordoquy, T.J. (2002) Shearinduced degradation of plasmid DNA. Journal of Pharmaceutical Sciences, 91, 1581-1589.
- Levy, M.S., O'Kennedy, R.D., Ayazi-Shamlou, P. and Dunnill, P. (2000) Biochemical engineering approches to the challenges of producing pure plasmid DNA. Trends on Biotechnology, 18, 296-305.doi:10.1016/S0167-7799(00)01446-3
- Frenkel, J. (1944) Orientation and rupture of linear macromolecules in dilute solutions under the influence of viscous flow. Acta Physicochima U.R.S.S., 19, 51-76.
- Bowman, R.D. and Davidson, N. (1972) Hydrodynamic shear breakage of DNA. Biopolymers, 11, 2601-2624.doi:10.1002/bip.1972.360111217
- Levinthal, C. and Davidson, P.F. (1961) Degradation of deoxyribonucleic acid under hydrodynamic shearing forces. Journal of Molecular Biology, 3, 674-683.doi:10.1016/S0022-2836(61)80030-2
- Dancis, B.M. (1978) Shear breakage of DNA. Biophysical Journal, 24, 489-503.doi:10.1016/S0006-3495(78)85396-X
- Adam, R.E. and Zimm, B.H. (1977) Shear degradation of DNA. Nucleic Acids Research, 4, 1513-1537.doi:10.1093/nar/4.5.1513
- Vanapalli, S.A., Ceccio, S.L. and Solomon, M.J. (2006) Universal scaling for polymer chain scission in turbulence. PNAS, 103, 16660-16665.doi:10.1073/pnas.0607933103
- Zhang, H., Kong, S., Booth, A., Boushable, R., Levy, M.S. and Hoare, M. (2007) Prediction of shear damage of plasmid DNA in pump and centrifuge operations using an ultrascale-down devices. Biotechnology Progress, 23, 858-865.
- Rock, C., Shamlou, P.A. and Levy, M.S. (2003) An automated microplate-based method for monitoring DNA strand breaks in plasmids and bacterial artificial chromosomes. Nucleic Acids Research, 31, e65.doi:10.1093/nar/gng065
- Hershey, A.D. and Burgi, E. (1960) Molecular homogeneity of the deoxyribonucleic acid of phage T2. Journal of Molecular Biology, 2, 143-152. doi:10.1016/S0022-2836(60)80016-2
- Burgi, E. and Hershey, A.D. (1962) Specificity and concentration limit in self-protection against mechanical breakage of DNA. Journal of Molecular Biology, 4, 313-315. doi:10.1016/S0022-2836(62)80009-6
- Levy, M.S., Collins, I.J., Yim, S.S., Ward, J.M., Titchener-Hooker, N., Shamlou, P.A. and Dunnil, P. (1999) Effect of shear on plasmid DNA in solution. Bioprocess Engineering, 20, 7-13. doi:10.1007/s004490050552
- Lim, H.-M., Yoo, H.-B., Hong, N.-S., Yang, I.-H, M.-S. and Park, S.-R. (2009) Count-based quantitation of macro-DNA molecules. Metrologia, 46, 375-387.doi:10.1088/0026-1394/46/3/028
- Manning, G.S. (1980) Thermodynamics stability theory for DNA doughnut shapes induced by charge neutralization. Biopolymers, 19, 37-59.doi:10.1002/bip.1980.360190104
- Simmonson, T. and Kubista, M. (1993) DNA orientation in shear flow. Bioploymers, 33, 1225-1235.doi:10.1002/bip.360330809