Journal of Biophysical Chemistry
Vol.2 No.4(2011), Article ID:8431,5 pages DOI:10.4236/jbpc.2011.24047
Structural analyses of the interactions of SoxY and SoxZ from thermo-neutrophilic Hydrogenobacterthermophilus
1Department of Biochemistry and Biophysics, University of Kalyani, Kalyani, India; *Corresponding Author: angshuman_bagchi@yahoo.com
2Bioinformatics Center, Bose Institute, Kolkata, India.
Received 22 July 2011; revised 2 September 2011; accepted 11 September 2011.
Keywords: Molecular Modeling; Sox Operon; Molecular Docking; Thermodynamics
ABSTRACT
Microbial redox reactions of inorganic sulfur compounds are one of the important reactions responsible for the recycling of this element to maintain the environmental sulfur balance. These reactions are carried out by phylogenetically diverse set of microorganisms. The sulfur oxidizing gene cluster (sox) of thermo-neutrophilic bacterium Hydrogenobacter thermophilus consists of soxYZAXB. The bacterium shows optimal thiosulfate oxidation activity at 60˚C. There are practically no reports regarding the structural biology of the sulfur oxidation process in this organism. In the present context, we employed homology modeling to construct the three dimensional structures of SoxY and SoxZ from Hydrogenobacter thermophilus. With the help of docking simulations we have identified the amino acid residues of these proteins involved in the interactions. The thermodynamics of the protein-protein interactions have also been analyzed. The probable biochemical mechanism of the binding of thiosulfate has been elucidated. Our study provides a rational framework to understand the molecular mechanism of the sulfur oxidation biochemistry.
1. INTRODUCTION
One of the major sets of biogeochemical reactions of our environment is the redox reactions of environmental sulfur compounds to maintain the balance of this important element in the atmosphere. Sulfur having a broad range of oxidation states, viz., +6 to –2, undergoes a number of important bioinorganic reactions which convert the sulfur from one form to the other. Such chemoor photolithotrophic reactions of sulfur are mediated by phylogenetically diverse set of microorganisms [1]. The natural sources of sulfur for these reactions are sulfide, thiosulfate, tetrathionate, etc., producing reductants which on the other hand are used for fixation of carbon di oxide or in the respiratory electron transfer chains [2]. The majority of the sulfur oxidizing-microorganisms are found to possess the gene cluster called the sox operon [3-10]. Nevertheless, the molecular mechanism of the sulfur oxidation process is poorly understood. The sox operon from mesophilic α-Proteobacteria like, Paracoccus pantotrophus (Para) and Rhodovulum sulfidophilum, contains two divergently transcribed transcriptional units comprising of genes soxSR and soxVWXYZABCDEFGH [3-10]. The proposed molecular mechanism of thiosulfate in Para shows that thiosulfate gets coupled to carboxy terminal Cysteine residue of SoxY complexed with SoxZ protein with the help of SoxXA. In the next step SoxB hydrolytically cleaves the SoxY-thosulfate adduct to release a sulfate ion. In Para, Sox(CD)2 and SoxB help to recycle the SoxY protein by hydrolytically releasing a sulfate from SoxY. In contrast to the mesophilic organisms there are practically no reports about the biochemistry of the sox operon of thermophilic microorganisms [10]. In the present scenario, we made an attempt to analyze the sulfur oxidation biochemistry of Hydrogenobacter thermophilus (Htherm). Htherm is a thermo neutrophilic organism and has been reported to be involved in the oxidation of thiosulfate to sulfate. Htherm possesses five sox genes, viz., soxYZAXB [10]. It has been observed that the thiosulfate oxidation activity of Htherm is temperature dependent and attains a peak at 60˚C [10]. However, the thiosulfate oxidation activity starts to decrease above the optimum temperature of 60˚C and is somewhat lost at 70˚C [10]. Since no structural analyses have been done with the sox operon of Htherm, we tried to explore the molecular biochemistry of thiosulfate oxidation of this organism. We used the amino acid sequences of SoxY and SoxZ of Htherm to build computational models of the respective proteins. We used molecular docking simulations in order to build a complex of SoxY and SoxZ, viz., the SoxYZ complex. The putative binding sites of SoxY and SoxZ were predicted and analyzed. In Para, sulfur anion (thiosulfate for example) combines with the carboxy terminal Cysteine (Cys 147) residue of SoxY and forms an adduct with the help of SoxZ protein [1,3-9]. However, to date the detailed structural information regarding the interactions between these proteins has not been fully understood. Though the molecular mechanism of the sulfur oxidation biochemistry is not well understood, these studies provide a detailed structural insight into the plausible molecular mechanism of the involvements of these proteins in the global sulfur oxidation reaction cycle. As this is the first report regarding the structural bases of the involvements of SoxY and SoxZ from Htherm in the process of biochemical oxidation of sulfur anions, our studies may contribute towards the understanding of the complex thiosulfate oxidation process in the thermoneutrophilic Htherm.
2. MATERIALS AND METHODS
2.1. Sequence Analysis and Homology Modeling of Monomeric SoxY and SoxZ Proteins
The amino acid sequences of SoxY and SoxZ proteins of Htherm were obtained from Entrez database (Accession no. NC_013799). To identify suitable templates for homology modeling the amino acid sequences of these proteins were used separately to search the Brookhaven Protein Data Bank (PDB) [11] using the software tools BLAST [12] and FUGUE [13]. The search results for SoxY and SoxZ picked up the X-Ray crystal structure of the SoxYZ complex from Paracoccus denitrificans (PDB Code: 2OXG; Y chain for SoxY and Z chain for SoxZ) with 35% and 45% sequence identities respectively. The Y and Z chains of 2OXG were separated and used to build the models of SoxY and SoxZ proteins of Htherm. The homology modeling was performed using the software suite present in the Homology module of Insight II (Accelrys, San Diego, CA, USA). The modeled structures were then superimposed separately on each of the crystal templates without altering the coordinate system of atomic positions in the respective templates (Y chain of 2OXG for SoxY, Z chain of 2OXG for SoxZ). The root mean square deviations (RMSD) for the superimpositions were 0.5Å for both SoxY and SoxZ. The mode of the superimposition of SoxY onto its crystal template was presented in Figure 1. Since the modes of superimpositions were similar for SoxZ only one (that of SoxY onto its crystal template) was presented. The models of the proteins were then energy minimized in two steps. In the first step, all the main and side chain atoms of the proteins were allowed to move and in the next step of the minimization process the backbones of the proteins were kept fixed and the side chains were allowed to move to ensure proper interactions. All energy minimizations were done using conjugate gradient [CG] with consistent valence force field (CVFF) [14] using the program DISCOVER until the structures reached the final derivative of 0.001 kcal/mole.
2.2. Validation of the Models
The Z-scores calculated using the software PROSA 2003 [15] showed that the predicted homology models were well inside the range of typical native structures [16]. The residue profiles of the three dimensional models were further checked by VERIFY3D [17]. PROCHECK [18] analyses were performed in order to assess the stereo-chemical qualities of the models and Ramachandran plots [19] were drawn. No residues were found to be present in the disallowed regions of the Ramachandran plots. All these methods predicted a good model quality.
2.3. Molecular Docking Simulations
In order to study the interactions between SoxY &

Figure 1. Superimposition of the α-carbon backbones of SoxY (Black) on 2OXG (Red).
SoxZ proteins as well as between the SoxYZ protein complex and thiosulfate, first the models of the SoxY and SoxZ proteins were docked using the software GRAMM [20]. The docking of the proteins was also performed with DOT [21] and ZDOCK [22], using the ClusPro server [23] in order to get a comprehensive result. The docked structure of the SoxYZ complex that yielded the best score was selected and analyzed visually using Insight II. The complex of SoxYZ was then energy minimized as per the protocol mentioned in the Sequence analysis and homology modeling of monomeric SoxY and SoxZ proteins section. The resulting energy minimized structure of the SoxYZ complex was used to dock with thiosulfate ion using the aforementioned software tools. The resulting best structure was again selected on the basis of the score and analyzed visually using Insight II. Again the SoxYZ-thiosulfate protein complex was subjected to energy minimization as per the protocol in the Sequence analysis and homology modeling of monomeric SoxY and SoxZ proteins section.
2.4. Molecular Dynamics Simulations
Molecular dynamics simulation of SoxYZ complex was performed using the GROMACS 3.3.1 package [24] and gmx (modified GROMOS 87) force field that is implemented on LINUX architecture. The SoxYZ protein complex was solvated in a triclinic water box having a dimension of 8.22 nm. The box contained approximately 10,000 SPC water molecules. All protein atoms were kept at a distance equal to or greater than 1.0 nm from edges of the box. In order to neutralize the charge of the system, requisite number of Na+ ions was added. The system was then subjected to energy minimization for 2000 steps by steepest descents (SD). The minimized system was then equilibrated for 50 ps at 333 K by position restrained molecular dynamics simulation to relax the solvent. The system after equilibration was subjected to molecular dynamics simulations for 20 ns at 333 K. The LINCS algorithm [25] was used for constraining bond lengths using 2 fs time step for all calculations. Particle Mesh Ewald (PME) [26] summation scheme was employed to calculate the electrostatic interactions. The cut-off used for van der Waals and Coulomb interactions was 0.9 nm. The non-bonded pair list was updated at an interval of 10 steps and conformations were stored for every 2 ps. The same protocol was used in order to perform the molecular dynamics simulations of SoxYZ protein complex with thiosulfate ion.
2.5. Calculation of Protein-Protein Interactions
To find out the interactions between the SoxY and SoxZ proteins, as well as between thiosulfate ion and the SoxYZ complex structural thermodynamics calculator software package [27] and the Biopolymer module of Insight II were used.
2.6. Thermodynamic Analyses of the Complexes
In order to find the mode of binding of the proteins and the binding of thiosulfate ion with the SoxYZ protein complex structural thermodynamics calculator (STC) [27] was used. The binding free energies of the SoxYZ complex and that of the soxYZ-thiosulfate complex were determined at temperatures ranging between 50˚C and 70˚C with an interval of 5˚C as SoxYZ complex of Htherm has an optimum temperature at 60˚C.
3. RESULT AND DISCUSSION
3.1. Description of the Structure of SoxY
The modeled structure of SoxY is a 148 amino acid residue long protein. The predicted structure is similar to the SoxY protein from the SoxYZ complex from Paracoccus denitrificans (PDB Code: 2OXG; Y chain for SoxY). The protein is composed mostly of β-sheets with an α-helix at the N-terminal end of the protein. At the middle of this protein there is a three-stranded anti-parallel β-sheet [amino acid residues 54 - 58, 68 - 74 and 109 - 114] which culminates in another three-stranded anti-parallel β-sheet (amino acid residues 83 - 89, 120 - 127 and 133 - 140) at the C-terminal end of the protein. The structure is presented in Figure 2.
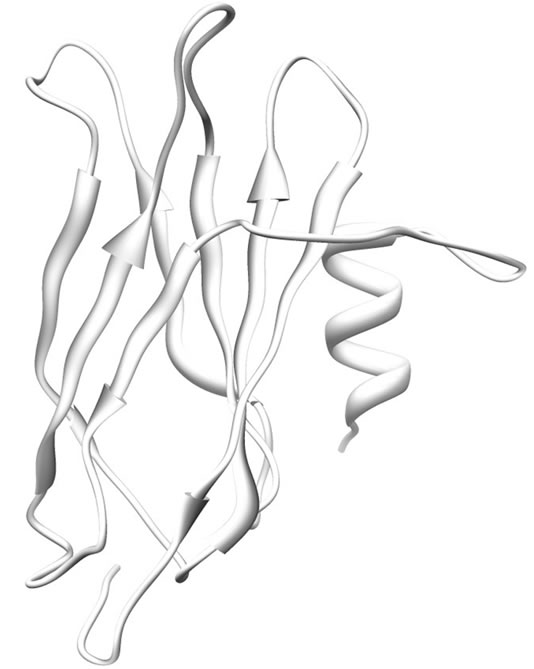
Figure 2. Ribbon representation of modeled SoxY. α-helices and β-sheets are shown as helices and sheets respectively. The rest are shown as loops.
3.2. Description of the Structure of SoxZ
The model of SoxZ protein consists of 109 amino acid residues. The predicted structure is similar to the SoxZ protein from the SoxYZ complex from Paracoccus denitrificans (PDB Code: 2OXG; Z chain for SoxZ). The protein is made up of β-strands [amino acid residues 5 - 11, 19 - 26, 49 - 57, 75 - 81, 86 - 94 and 99 - 106], which produce a six-stranded anti-parallel β-sheet connected by loops. Figure 3 shows the structure of the modeled protein.
3.3. Interaction of SoxY with SoxZ
SoxY and SoxZ are found to interact strongly with each other. The protein-protein interface is found to mainly contain the polar amino acid residues. The interior of the complex is made up of hydrophobic amino acids. There are extensive H-bonding interactions involving both the main and the side chains of the two protein molecules. The SoxYZ complex is also stabilized by ionic interactions. SoxY protein of Htherm contains a sulfur anion binding signature GGCGG sequence at the carboxy terminal region of the protein where the free thiol group of the Cysteine residue 147 binds the sulfur anion (thiosulfate). Interestingly, amino acid residues from the sulfur anion binding motif of SoxY do not take part in this interaction scheme. The sulfur anion binding
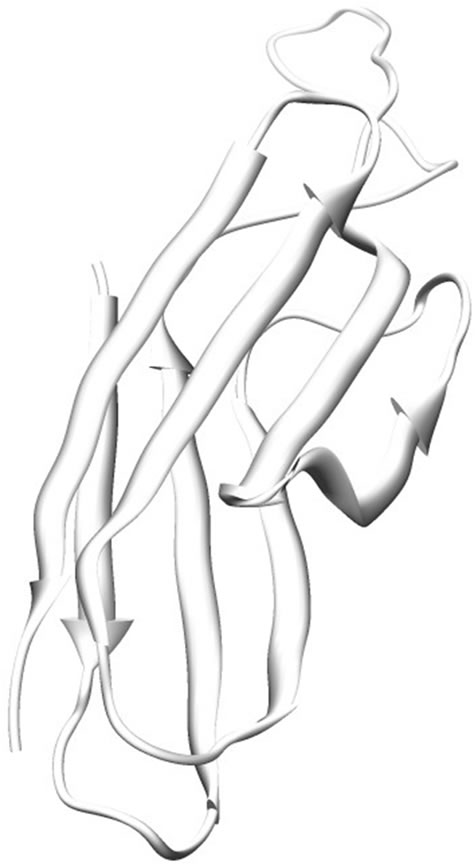
Figure 3. Ribbon representation of modeled SoxZ. β-sheets are shown as sheets respectively. The rest are shown as loops.
region of SoxY forms a cavity which is surrounded by 5 positively charged amino acid residues (Lys36, Lys51, Lys59, Lys81 and Lys89) of SoxZ. This positive environment in the complex may help to drag the sulfur anion near the active site of SoxY which then covalently binds the anion. Figure 4 represents the binding of the two proteins. The active site Cysteine residue (Cys147) is found to be freely available in the SoxYZ complex. The other positively charged amino acid residues (mainly Lys12, Lys16, Lys17 and Lys21) from SoxZ also helped in the process of binding of the sulfur anions.
3.4. Thermodynamics of the Interactions
The change in free energy values of the SoxYZ and the SoxYZ-thiosulfate complexes were measured with STC. The changes in free energy values for the SoxYZ protein complex are presented in Table 1. It is clear from the table that the binding energies of the interactions between the proteins are maximum at 60˚C and decreases gradually after 60˚C and becomes the minimum at 70˚C. Thus, it can be concluded that the proteins interact optimally at 60˚C.
4. CONCLUSIONS
In this paper an attempt is made to identify the structural basis of involvement of SoxY and SoxZ in the bio-
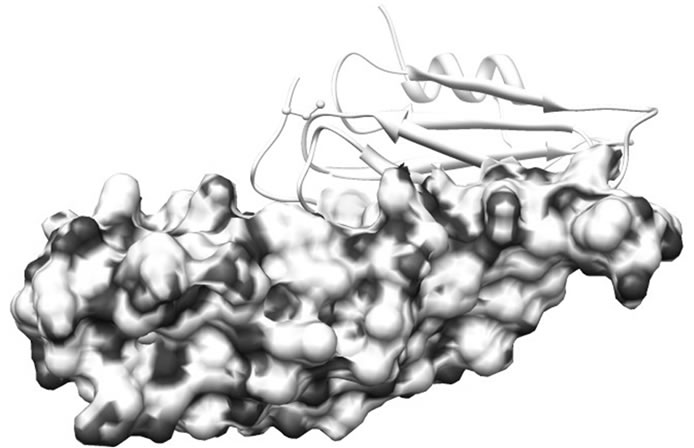
Figure 4. Electrostatic surface charge distribution of SoxYZ complex. The thiosulfate binding region of SoxY is surrounded by electrostatically positive regions of SoxZ.
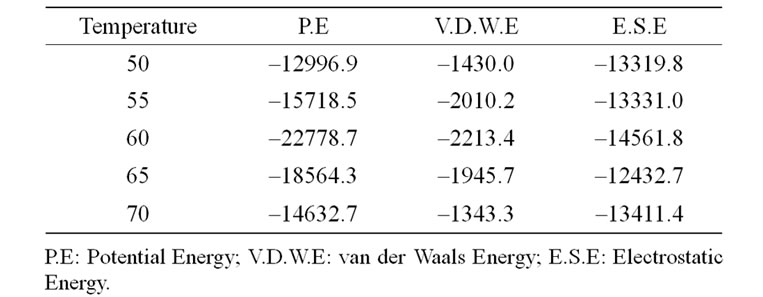
Table 1. Variation of the different energy terns in different temperatures in Kcal/mole.
chemical oxidation of thiosulfate in Htherm. We have described and analyzed the three-dimensional structures of SoxY and SoxZ. We also established the geometry of the sulfur anion binding region of SoxY. The putative mode of binding of SoxY with SoxZ was analyzed and the possible molecular basis of binding of thiosulfate has been described. The plausible molecular biology for the formation of the heterodimeric complex of SoxYZ has also been demonstrated to predict the biochemical pathway of sulfur anion binding and oxidation reaction cycle as the SoxYZ complex is the central player in the oxidation of sulfur anions. The thermodyanamics of the binding of SoxY and SoxZ have also been demonstrated. Since there have been no previous reports regarding the structural biology of these proteins, results from this study may shed light to understand the three dimensional structures of SoxY and SoxZ as well as to elucidate the structural basis of the molecular functions of these proteins. Our model provides a rational framework for designing experiments to determine the contribution of the various amino acid residues in these proteins to predict the molecular basis of their interactions both among themselves as well as with various sulfur anions.
5. ACKNOWLEDGEMENTS
The valuable comments from the anonymous reviewers are greatly acknowledged. The authors would like to acknowledge the technical help from the DBT, India sponsored Bioinformatics Infrastructure Facility of University of Kalyani.
REFERENCES
- Freidrich, C.G. (1998) Physiology and genetics of sulfuroxidizing bacteria. Advances in Microbial Physiology, 39, 235-289. doi:10.1016/S0065-2911(08)60018-1
- Le Faou, A., et al. (1990) Thiosulfate, polythionates and elemental sulfur assimilation and reduction in the bacterial world. FEMS Microbiology Reviews, 6, 351-381. doi:10.1016/S0168-6445(05)80005-1
- Appia-Ayme, C., et al. (2001) Cytochrome complex essential for photosynthetic oxidation of both thiosulfate and sulfide in Rhodovulum sulfidophilum. Journal of Bacteriology, 183, 6107-6118. doi:10.1128/JB.183.20.6107-6118.2001
- Freidrich, C.G., et al. (2001) Oxidation of reduced inorganic sulfur compounds by bacteria: Emergence of a common mechanism? Applied and Environmental Microbiology, 67, 2873-2882. doi:10.1128/AEM.67.7.2873-2882.2001
- Bagchi, A. and Ghosh, T.C. (2006) Structural insight into the interactions of SoxV, SoxW and SoxS in the process of transport of reductants during sulfur oxidation by the novel global sulfur oxidation reaction cycle. Biophysical Chemistry, 119, 7-13. doi:10.1016/j.bpc.2005.08.011
- Bagchi, A., et al. (2005) Homology modeling of a transcriptional regulator SoxR of the lithotrophic sulfur oxidation (Sox) operon in α-proteobacteria. Journal of Biomolecular Structure & Dynamics, 22, 571-578.
- Bagchi, A. and Roy, P. (2005) Structural insight into SoxC and SoxD interaction and their role in electron transport process in the novel global sulfur cycle in Paracoccus pantotrophus. Biochemical and Biophysical Research Communications, 331, 1107-1103. doi:10.1016/j.bbrc.2005.04.028
- Rother, D. and Friedrich, C.G. (2002) The cytochrome complex SoxXA of Paracoccus pantotrophus is produced in Escherichia coli and functional in the reconstituted sulfur-oxidizing enzyme system. Biochimica et Biophysica Acta, 1598, 65-73.
- Hensen, D., et al. (2006) Thiosulphate oxidation in the phototrophic sulphur bacterium Allochromatium vinosum. Molecular Microbiology, 62, 794-810. doi:10.1111/j.1365-2958.2006.05408.x
- Sano R., et al. (2010) Thiosulphate oxidation by a thermo-neutrophilic hydrogen-oxidizing bacterium, Hydrogenobacter thermophilus. Bioscience, Biotechnology, and Biochemistry, 74, 892-894. doi:10.1271/bbb.90948
- Berman, M.H., et al. (2000) The protein data bank. Nucleic Acids Research, 28, 235-242. doi:10.1093/nar/28.1.235
- Altschul, S.F., et al. (1990) Basic local alignment search tool. Journal of Molecular Biology, 25, 403-410.
- Shi, J., et al. (2001) FUGUE: Sequence-structure homology recognition using environment-specific substitution tables and structure-dependent gap penalties. Journal of Molecular Biology, 310, 243-257. doi:10.1006/jmbi.2001.4762
- Dauber-Osguthorpe P., et al. (1988) Structure and energetics of ligand binding to proteins: Escherichia coli dihydrofolate reductase trimethoprim, a drug receptor system. Proteins, 4, 31-47. doi:10.1002/prot.340040106
- Sippl, M.J. (1993) Recognition of errors in three-dimensional structures in proteins. Proteins, 17, 355-362. doi:10.1002/prot.340170404
- Wiederstein, M., et al. (2004) Evolutionary methods in Biotechnology, Wiley-VCH.
- Eisenberg, D., et al. (1997) VERIFY3D: Assessment of protein models with three-dimensional profiles. Methods in Enzymology, 277, 396-404. doi:10.1016/S0076-6879(97)77022-8
- Laskowski, R.A., et al. (1993) PROCHECK: A program to check the stereochemistry of protein structures. Journal of Applied Crystallography, 26, 283-291. doi:10.1107/S0021889892009944
- Ramachandran, G.N. and Sashisekharan, V. (1968) Conformation of polypeptides and proteins. Advances in Protein Chemistry, 23, 283-438. doi:10.1016/S0065-3233(08)60402-7
- Vakser, I.A. (1995) Protein docking for low-resolution structures. Protein Engineering, 8, 371-377. doi:10.1093/protein/8.4.371
- Mendel, J.G., et al. (2001) Protein docking using continuum electrostatics and geometric fit. Protein Engineering, 14, 105-113. doi:10.1093/protein/14.2.105
- Chen, R., et al. (2003) ZDOCK: An initial-stage protein docking algorithms. Proteins, 51, 82-87.
- Comeau, S.R., et al. (2004) ClusPro: An automated docking and discrimination method for the prediction of protein complexes. Bioinformatics, 20, 45-50. doi:10.1093/bioinformatics/btg371
- van der Spoel, D., et al. (2005) GROMACS: Fast, flexible and free. Journal of Computational Chemistry, 26, 1701-1718. doi:10.1002/jcc.20291
- Hess, B., et al. (1997) LINCS: A linear constraint solver for molecular simulations. Journal of Computational Chemistry, 18, 1463-1472. doi:10.1002/(SICI)1096-987X(199709)18:12<1463::AID-JCC4>3.0.CO;2-H
- Essmann, U., et al. (1995) A smooth particle mesh Ewald method. Journal of Chemical Physics, 105, 8577-8593. doi:10.1063/1.470117
- Lavigne, P., et al. (2000) Structure-based thermodynamic analysis of the dissociation of protein phosphatase-1 catalytic subunit and microcystin-LR docked complexes. Protein Science, 9, 252-264. doi:10.1110/ps.9.2.252