Journal of Cancer Therapy
Vol.10 No.02(2019), Article ID:90650,12 pages
10.4236/jct.2019.102013
Sequential and Dual Inhibition of Pleiotropic Targets in Cancer―A Novel Strategy to Sensitize Tumor Cells to Targeted Therapies and Overcome Resistance
M. Nezami
Pacific Medical Center of Hope and Research Cancer Institute of America, Fresno, CA, USA

Copyright © 2019 by author(s) and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY 4.0).
http://creativecommons.org/licenses/by/4.0/



Received: January 21, 2019; Accepted: February 18, 2019; Published: February 21, 2019
ABSTRACT
In this paper we discuss the rationale of applying a “sequential” targeted therapy with a specific application in clinical practice, given our understanding of cancer heterogenous and dynamic biology. We explore the advantages of “single inhibition” to combinational therapies and dual inhibition on key pathways, as well as a multi-step approach to use “oncological addiction” and “oncogenic shock” as a suicide plan for cancer. We specifically explain how the downstream targets can be used to “create” feedback loops in an advantage for creating actionable targets in upstream signaling molecules. We apply this hypothesis in the clinical setting, with superior outcomes shown in a series of case studies. We conclude that “sequential and dual inhibition” can be considered a meaningful approach to checkmate the tumor, with minimum chance of tumor resistance. We recommend further clinical studies to generate further hypotheses based on each actionable target.
Keywords:
Targeted Therapies, Multitargeted Epigenetic Therapy, Oncogenic Addiction, Tumor Resistance, Tyrosine Kinase

1. Background
It is well described in the literature that oncogenic driver events decisively influence the viability and clinical behavior of a given tumor [1] . Some of the driver mutations are found to be targets for therapy, whereas others play crucial roles in resistance to therapy. The challenge is that tumor cells have a very sophisticated mechanism of survival, by switching their driving pathways and signaling transduction pathways in a dynamic fashion. This understanding has prompted efforts aimed at treating tumor cells with multiple drugs to inhibit as many targets as possible, simultaneously [2] . The majority of combinational therapies have been designed around the understanding of this phenomenon; however, clinical advantages of combinational therapies have been limited, and many of the trials have shown minimum or no clinical benefit of such approach.
Tumor cells mainly grow when there is over expression, augmentation, or activation of mutations present in two key pathways: PI3k/mTOR, and EGFR/KRAS pathways. Unfortunately, as much as we realize that the inhibition of the upstream molecules is necessary for blockade of the growth, we do not yet have effective therapies that target KRAS/NF-1 or PI3k. The targeted therapies primarily treat and target downstream molecules, leaving the feedback loops intact, which creates further resistance. We also know that most drugs that target PI3k or KRAS are extremely toxic or ineffective [3] [4] .
A few well-studied targeted therapies are drugs that block EGFR, HER-2, and BRAF. Only a minority of tumor cells in certain types of tumors harbor an effective target for EGFR blocking agents, such as small frame deletions in EGFR exon 19 or exon 21 L858R mutation. Therefore, if the EGFR target is not over expressed, the inhibition is ineffective. For the small group of patients with actionable EGFR targets, tumor resistance becomes a challenge [5] [6] . Secondary mutations in the EGFR target are seen in 50% of cases (such as exon 20 T790M). Activation of KRAS, PI3k and c-Met (HGF) may also occur, making these tumors resistant after a period of time [7] . As far as the HER-2 target, only 30% of tumors clinically respond to the drug trastuzumab, due to alternative pathways, and tumors harboring ERBB2 overexpression develop resistance quickly [8] . The same is true for the BRAF target. In the case of colorectal cancer with mutated BRAF, the inhibition may not be clinically beneficial, due to the activation of KRAS, c-Kit or ALK expression (as escape pathways) [9] . ALK can stimulate BRAF, and vice versa.
To overcome such failures, scientists have suggested using a multi-tyrosine kinase inhibitor as a multi-targeted approach. One example would be the use of selumetinib or ragorafenib, which target both angiogenesis as well as different key pathway targets, or combinational therapy [10] . Although it was apparent that inhibition of EGFR and BRAF has synergistic effects in vitro and in vivo in subset of BRAF mutated colon CA, the clinical advantage was not convincing [11] . Results of such therapies have been clinically disappointing in the majority of cases. This approach has also used dual inhibition unsuccessfully, such as combining trametinib with everolimus in melanoma or other solid tumors or combining everolimus with erlotonib in pancreatic cancer [12] . Unfortunately, the results have been discouraging, as well as causing a great amount of toxicity with this combinational approach. The current research is still on-going on approval of dual pathway blockers, such as inhibitors of Akt and HER 3 [13] [14] .
Our understanding of the cancer network of cross talks has become increasingly complicated. The problem arises specially when this cross talk is dynamic, and it can adjust to either single targeted therapy or a combination of targets when the treatment is applied as a “static”, or one step approach.
The evolutionary trajectory of spontaneous human tumors explains the dynamic process for driver designation of a tumor. To our knowledge, it has never been suggested or explored to treat tumor biology and its heterogenous character as a moving target, with an adaptive strategy that could adjust to tumor response or resistance. Our knowledge of modern sequencing of tumor DNA enables us to look at the point mutations (as well as the copy numbers, and chromosomal alterations) and has prompted us to further explore a “roadmap” to create a plan of attack based on the tumor’s driver status, and the sequential strategy of inhibition, which we discuss here in detail.
In this approach, instead of using combination therapy which has been claimed to improve patient outcomes, by definition, we create an oncological addiction. By inhibiting single downstream targets, we keep the doors open for escape pathways.
“Oncogenic addiction”, a term first described a decade ago by Weinstenand further proved as a meaningful therapeutic strategy for the first time, in CML (BCR-ABL), proposes a single dominant oncogene for growth and survival [15] .
Despite the presence of heterogeneity (diverse array of genetic lesions), the inhibition of such driver, could translate to halt the neoplastic phenomenon, as long as the mechanisms of resistance are explored and effectively treated. In our model, the PI3k and IGF-1 pathway were identified and treated as parallel transduction pathways to overcome resistance. Further, we reduce the genomic instability of a tumor by epigenetic modification of gate keeper and caretaker oncosuppresors, therefore inhibiting the tumor’s tendency to switch its driver.
In our model of treatment, the initial stage of the therapy would consist of treating a target that is effectively inhibited, thus creating “maximum” feedback loops. In this initial stage we see benefit from a biologic phenomenon described as “Genetic Streamlining” to induce addiction. By doing so, we create “genomic degeneration”, which is mutational burden of non-adaptive (passive) alterations. In this situation, the tumor is suffering from replication stress. The selective pressure caused by the drugs creates the addiction, which we take advantage in the second stage. The second stage of the therapy initiates after feedback loops are completely activated, and the tumor selectively starts to use the streamline. This stage inhibits the driver, a process which by reversing oncogenic shock reverses the disengagement of proapoptotic safeguards.
2. Methods and Materials
The network of cross talk in RAS/RAF/Mek/Erk/MAPK was targeted by selective inhibition at lowest downstream transduction, as shown in Image 1.
In Image 2, the cross talk of alternative pathways is simply illustrated through Pi3k pathway. This alternative pathway would be used, and would be sequentially be targeted, after its activation.
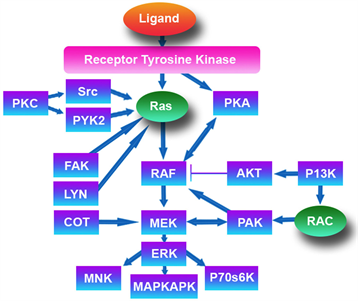
Image 1. MAPK pathway.

Image 2. PI3K cross talks.
In the first stage, we choose Erk/ MAPK/HDAC and cMyc targets, and we selected several different agents to inhibit them. The Erk target was selected because the process of transcription, differentiation, survival, and proliferation are all dependent on Erk/ELK activation, as is the cell adhesions through integrins (see Image 3 and Image 4).
As illustrated in Image 3, HDAC and cMyc inhibition is critical in activation of the feedback loops through Erk. Our findings have revealed a strong inhibition of cMyc in all cases treated by this protocol. We have shown complete eradication of cMyc positive circulating tumor cells (CTCs) in all samples with this protocol. We also profiled tumors and identified presence of lack of Pi3K pathway activation. We used tissue and liquid biopsies to show this. We also identified the most relevant upstream molecules to be targeted at each point of time depending on the dynamic presence of circulating tumor DNA. With this approach, we could select the exact upstream target that was overexpressed due to our initial step of therapy, to be inhibited. We aimed to correlate the clinical outcome with our approach and use historical data to compare results in single patients. We were able to show “significant applicable” clinical advantage of this approach, as compared to the standard of care, in a setting of Stage IV and refractory disease. The compounds we used to inhibit HADC and Erk included Sodium Phenylbutyrate and Quercetin, in a protocol named multitargeted epigenetic therapy or MTET. This protocol sensitized and synergized with targeted therapies we used in these series, Trametinib as an example. In other cases, we inhibited MAPK with an aromatase inhibitor, or Lupron. The compounds we used to inhibit EGFR were erlotonib ortrastuzumab (for ERBB2), and the compound we used to inhibit Mek was trametinib. For PI3k we used riluzole (by inhibition of coupling of PI3k with NMDA).
It is important to reemphasize that patients’ laboratory findings as well as imaging were used to study response to each step of therapy. In initial stage when the Erk/MAPK/HDAC/and c Myc is inhibited we would generally observe a stable disease identified through surrogate markers or scans, after a while the feedback loops would have been activated (intentionally) by our therapy, making the tumor grow and bypass the inhibition of MAPK/Erk. (The known feedback loops between Erk and Mek and between Erk and RAF has been studied).
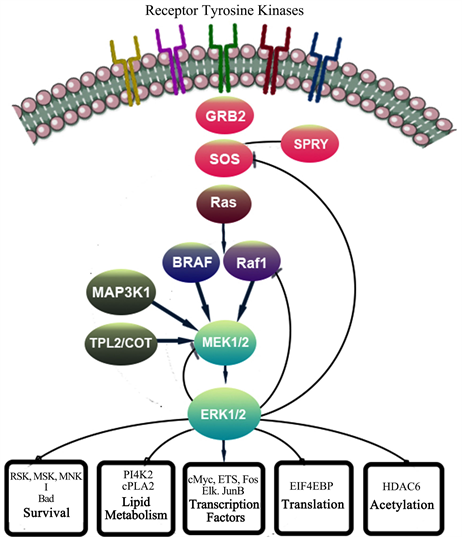
Image 3. Erk Cross talks and post translational modifications.
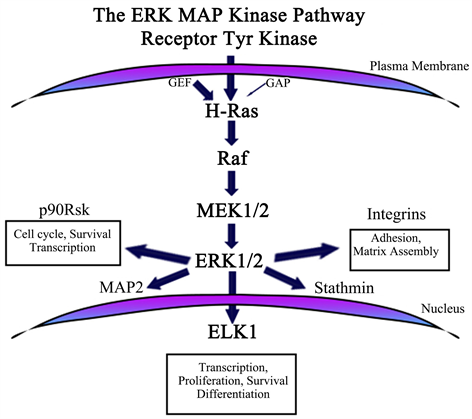
Image 4. Erk Cross talks and post translational modifications.
We identified the surrogates of activated feedback loops through increased mutation allele fraction (MAF) of circulatory DNA for RAF, NF-1, KRAS, PI3k, OR increased serum levels of HER-2 in patients with Her 2 positive disease.
At this stage we would switch the therapy or add an upstream target inhibition. The time interval for stage one is two to eight week (two months). Further we measure (indirectly) oncogenic addiction by surrogate markers as companion diagnostics, and we apply the second stage therapy. The second stage therapy will inhibit the driver pathways effectively measured again by markers and imaging.
Case Studies:
Case 1: A 68-year-old female presented to our clinic for evaluation and management in 2014. She had a history of breast cancer (stage unknown), but no LNs involved, diagnosed in 2001. She was status post lumpectomy and hormonal therapy (which consisted of letrozole for 3 years andanastrazole for 4 years), and radiation. She had a recurrence of the tumor in same side documented in September 2013. She had a biopsy and was referred for mastectomy, and possible chemotherapies based on a PET scan her oncologist has ordered. She underwent care for her recurrence at Stanford, where she was treated with AC chemo regimen, while at the same time, receiving epigenetic treatments with our clinic. After chemotherapy was completed, we prescribe Lupron and Tamoxifen, as well as treated her with IV Quercetin. During this time we monitored her imaging and ctDNA. Her imaging revealed no evidence of disease, while her ctDNA showed positive NF-1, then disappears or NF-1 after treatment (see Figure 1).
She continues to be in remission and seen for maintenance therapies.

Figure 1. Pre and Post therapy c DNA.
Case 2: A 37-year-old female presented to our clinic for evaluation and management in 2015. She had a history of breast cancer diagnosed in 1/2014, stage IIA, with ER/ PR/HER-2 positive. At the time of diagnosis, she refused all conventional therapies, including hormonal blockade, opted for alternative treatments in Mexico. She exhausted IPT, Vit C drip and other alternative treatments, including autologous immune cell therapy and hyperthermia, at clinic in Mexico, and is referred to us by her physician in Ohio. She was status post mastectomy, which was done 1/2014, and recurrence of the tumor in 9/2015 at the surgical implant replacement. She has been followed by Cleveland clinic. Her scan in November 15, prior to her first presentation to our clinic, confirmed presence of innumerous bony mets as well as thoracic and neck LN involvements, extensive chest and bone disease, Stage IV metatstatic disease.
She was treated by our therapies with some success, as her PET scans showed improvement. She was started on trastuzumab, based on HER-2 + CTCs and elevated serum HER-2. HER-2 disappeared on ctDNA, which became completely undetectable, along with imaging showing regression of disease. She continues to be treated at our clinic (see Figure 2).
Case 3: A 51-year-old male presented to our clinic for evaluation and management. He had a history of Stage II melanoma that was diagnosed in 2004, after a mole on his right thumb was biopsied and proved to be melanoma. At the time he received no treatments, and his cancer recurred in January of 2015. in his right axilla. In November 2015 he underwent axillary lymph node dissection, and had 28 lymph nodes removed, 14 of which were positive for melanoma. He then received Virotherapy (RigVir treatments), and yet his cancer was progressed, as documented on 3/25/16 with a PET scan that showed extensive involvement of his right axillary and subpectorallymph nodes, as well as subcutaneous nodules, and right upper back, right supraclavicular fossa, Right upper lobe of the lung and septum. Upon his arrival he was assessed through liquid biopsies and tissue molecular profiling using whole genome next generation sequencing. His liquid biopsy revealed ctDNA which was positive for NF-1 at MAF of 0.7%. His molecular profiling of his tumor through Caris, showed positive NF-1 mutation at a different exon, as well as positive EGFR at exon 18, with Y727Y mutation. His circulatory tumor cells (CTC) were also turned out to be positive for cKIT prior to the treatments. His PDL-1 was negative. To our knowledge again this is the first case of melanoma with both NF-1 and EGFR mutations reported in the literature. His labs also showed elevated absolute eosinophil count at 0.6 percent. S100 B elevated at 120 (normal less than 96), and IGF-1 of 222 prior to the treatments, on 4/15/16. The options discussed with the patients and he decided to opt out of his oncologist recommendation to start Keytruda as a PD1 inhibitor. He immediately started the treatments with us and was treated using the MTET protocol (sodium phenyl butyrate and Quercetin) to inhibit theErk/Histone deacetylase (HDAC) and cMyc, as initial stage of his treatment.
His labs were repeated, and it showed his CTC showed no expression of cKIT, which to our knowledge is a dismal finding in melanoma with no available therapies. His CTC switched from using alternative pathways of metabolism of mitochondria to telomerase. His CBC showed normalization of eosinophils as a sign of reduced angiogenesis. Further his NF-1 MAF at ctDNA level increased to 2.1 percent, indicating the possibility of oncogenic addiction and activation of feedback loops. Then he was started on trametinib to inhibit the over-activated MEK. This stage was started on 6/30 with extraordinary results, as described here. His c DNA dropped to 0.2 percent from 2.1 (please see Figure 3).
His PET scan showed on 8/1/16 that he achieved a complete remission with

Figure 2. Pre and Post therapy c DNA.

Figure 3. Pre and Post therapy c DNA.
resolution of all his original and metastatic lesions (axillary, subpectoral, supraclavicular, upper back and left abdominal wall, left lower extremities, intraatrium septum in the heart, cervical, pulmonary, right upper lobe, groin, …) compared to 3/16 scan. His S 100 B came down to 70 measured on 8/30/16 (from 432). Further, it dropped down to 53 on 11/1/16.
His PET scan was repeated on 11/3/16 and it showed complete resolution of subcentimeter nodule in his leg, with near complete resolution of all metastatic disease. Since this patient had not carried mutated BRAF gene, the response to the combination of Trametinib and epigenetic protocol (MTET) was unexpected. It is noticeable that his maximum response was achieved with the combination of such therapies.
3. Conclusion
Our knowledge on oncogenic addiction gaining full validity has paradigmatically shown immediate translational relevance. We here conclude that a dynamic and adaptive approach to target the most dependent pathways in tumor is essential to a successful strategy. Further, to maximize the effectiveness of such inhibition, “creation” of dependency in tumor cells is achieved by an iatrogenic oncological addiction through downstream molecules inhibition. This to our knowledge is revolutionary as it “induces” feedback loops to “prepare” tumor for second phase inhibition. Our preliminary findings confirm that such sequential approach is viable and superior to the historical clinical outcome specially compared with combinational targeted approach in variety of tumor types and deserves further attention and applications in clinical trials.
Conflicts of Interest
The author declares no conflicts of interest regarding the publication of this paper.
Cite this paper
Nezami, M. (2019) Sequential and Dual Inhibition of Pleiotropic Targets in Cancer―A Novel Strategy to Sensitize Tumor Cells to Targeted Therapies and Overcome Resistance. Journal of Cancer Therapy, 10, 166-177. https://doi.org/10.4236/jct.2018.102013
References
- 1. Tuna, M. and Amos, C.I. (2012) Activating Mutations and Targeted Therapy in Cancer. Mutations in Human Genetic Disease, Chapter 14, 273-292. https://doi.org/10.5772/48701
- 2. Lu, J.-J., Pan, W., Hu, Y.-J. and Wang, Y.-T. (2012) Multi-Target Drugs: The Trend of Drug Research and Development. PLoS ONE, 7, e0040262. https://doi.org/10.1371/journal.pone.0040262
- 3. Chia, S., Gandhi, S., Joy, A., Edwards, S., Gorr, M., Hopkins, S. and Dent, S. (2015) Novel Agents and Associated Toxicities of Inhibitors of the pi3k/Akt/mtor Pathway for the Treatment of Breast Cancer. Current Oncology, 22, 33. https://doi.org/10.3747/co.22.2393
- 4. https://am.asco.org/exploring-pathway-pi3k-pathway-fact-sheet
- 5. Stewart, E.L., Tan, S.Z., Liu, G. and Tsao, M.S. (2015) Known and Putative Mechanisms of Resistance to EGFR Targeted Therapies in NSCLC Patients with EGFR Mutations—A Review. Translational Lung Cancer Research, 4, 67-81.
- 6. Cortot, A.B. and Janne, P.A. (2014) Molecular Mechanisms of Resistance in Epidermal Growth Factor Receptor-Mutant Lung Adenocarcinomas. European Respiratory Review, 23, 356-366. https://doi.org/10.1183/09059180.00004614
- 7. Misale, S., Yaeger, R., Hobor, S., Scala, E., Janakiraman, M., Liska, D., Valtorta, E., Schiavo, R., Buscarino, M., Siravegna, G., Bencardino, K., Cercek, A., Chen, C.T., Veronese, S., Zanon, C., Sartore-Bianchi, A., Gambacorta, M., Gallicchio, M., Vakiani, E., Boscaro, V., Medico, E., Weiser, M., Siena, S., Di Nicolantonio, F., Solit, D. and Bardelli, A. (2012) Emergence of KRAS Mutations and Acquired Resistance to Anti-EGFR Therapy in Colorectal Cancer. Nature, 486, 532-536. https://doi.org/10.1038/nature11156
- 8. Luque-Cabal, M., García-Teijido, P., Fernández-Pérez, Y., Sánchez-Lorenzo, L. and Palacio-Vázquez, I. (2016) Mechanisms behind the Resistance to Trastuzumab in HER2-Amplified Breast Cancer and Strategies to Overcome It. Clinical Medicine Insights. Oncology, 10, 21-30. https://doi.org/10.4137/CMO.S34537
- 9. Villanueva, J., Vultur, A. and Herlyn, M. (2011) Resistance to BRAF Inhibitors: Unraveling Mechanisms and Future Treatment Options. Cancer Research, 71, 7137-7140. https://doi.org/10.1158/0008-5472.CAN-11-1243
- 10. Yardley, D.A. (2013) Drug Resistance and the Role of Combination Chemotherapy in Improving Patient Outcomes. International Journal of Breast Cancer, 2013, 1-15. https://doi.org/10.1155/2013/137414
- 11. Nagaraja, A.K. and Bass, A.J. (2015) Hitting the Target in BRAF-Mutant Colorectal Cancer. Journal of Clinical Oncology, 33, 3990-3992. https://doi.org/10.1200/JCO.2015.63.7793
- 12. Tolcher, A.W., Bendell, J.C., Papadopoulos, K.P. and Infante, J.R. (2014) A Phase IB Trial of the Oral MEK Inhibitor Trametinib (GSK1120212) in Combination with Everolimus in Patients with Advanced Solid Tumors. Annals of Oncology, 26, 58-64. https://doi.org/10.1093/annonc/mdu482
- 13. Chandarlapaty, S., Sawai, A., et al. (2011) AKT Inhibition Relieves Feedback Suppression of Receptor Tyrosine Kinase Expression and Activity. Cancer Cell, 19, 58-71.
- 14. Tao, J.J., Castel, P., Radosevic-Robin, N., Elkabets, M., Baselga, J. and Scaltriti, M. (2014) Antagonism of EGFR and HER3 Enhances the Response to Inhibitors of the PI3K-Akt Pathway in Triple-Negative Breast Cancer. Science Signaling, 7.
- 15. Torti, D. and Trusolino, L. (2011) Oncogene Addiction as a Foundational Rationale for Targeted Anti-Cancer Therapy: Promises and Perils. EMBO Molecular Medicine, 3, 623-636. https://doi.org/10.1002/emmm.201100176