International Journal of Organic Chemistry
Vol.08 No.03(2018), Article ID:87651,16 pages
10.4236/ijoc.2018.83024
Imidoyl Radicals for the Synthesis of Chromenoquinolines and Naphthyridines
Karen C. Vizcaíno-Escobedo, J. Adrián Sánchez-Carrillo, Martha Albores-Velasco*
Facultad de Química, Universidad Nacional Autónoma de México, Ciudad Universitaria, Ciudad de México, México
Copyright © 2018 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY 4.0).
http://creativecommons.org/licenses/by/4.0/



Received: July 15, 2018; Accepted: September 27, 2018; Published: September 30, 2018
ABSTRACT
The stability of imidoyl radicals prepared from 2-(2-alkenyloxy)-phenyl selenoimidates and 2-(2-allylamino)-phenyl selenoimidates and their intramolecular free radical cyclization through the slow addition of tri-N-butyltin hydride and AIBN to obtain 4-chromanones and 4-quinolones were studied. The extension of the methodology to N-phenyl substituted imidoyl selenoates produced tandem cyclizations of the imidoyl radical with the allyl substituent, followed for a second cyclization of the intermediate radical with the aromatic substituent to produce dihydro-chromenoquinolines or tetrahydro-dibenzonaphtyridines. Further oxidation with DDQ produced: 6H-chromeno [4,3,b] quinoline and 7-methyl-6H-chromenoquinoline from 2-alkenylamino-phenylbenzamide and 5,6-dihydro-dibenzo [b,h][1,6] naphthyridine and 5,6-dihydro-7-methyl-dibenzo [b,h][1,6] naphthyridine from 2-2-(alkylamino)-N-phenylbenzamides. The cyclization of 2-(N,N-di-prop-2.enylamino)-N-phenylnicotinamide selenoate obtained from 2-aminonicotinic acid, produced 5,6-dihidro-benzo[b]pyrido2,3-h] [1,6] naphthyridine.
Keywords:
Imidoyl Radicals, Quinolones, Chromenoquinolines, Naphthyridines, Dibenzonaphtyridines

1. Introduction
Chromanones, quinolones, chromenoquinolines and naphthyridines constitute important polycycles of pharmaceutic antibacterial compounds; they are also present in natural products. The search of new synthetic method for these heterocycles can contribute to the development of new and better pharmaceutic compounds.
In this work, we present the synthesis of chromanones and quinolones by a radical intramolecular cyclization of O-allyl or N-allyl phenyl selenoimidates, prepared from imidoyl chlorides.
The extension of this reaction to aromatic substituted selenoimidates gave chromenoquinolines and benzonaphtyridines through a tandem cyclization of the imidoyl radical and the allyl substituent followed of a second cyclization with the aromatic substituent and further oxidation of the dihydro or tetrahydro polycycles.
Imidoyl radicals, isoelectronic to acyl radicals, are mainly obtained from alkyl-radical addition to isocyanides: Curran and Liu [1] prepared heterocyclic compounds from 5-iodo-1-pentine with an excess of phenyl isonitrile, AIBN and tributyltin hydride to obtain 2,3-dihidro-1H-cyclopenta [b] quinoline, Nanni et al. [2] [3] obtained 2,4-disubstituted quinolines and Zanardi et al. [4] prepared pyrido-2,4-triazines and quinolines from the reaction of arylideneaniline with diisopropyl-peroxydicarbonate and phenyl acetylenes and proposed the formation of α-(arylthio)-imidoyl radicals.
Uneyama et al. [5] obtained indoles from an intramolecular photochemical homolysis of azo compounds derived of imidoyl iodides to triple bonds and Zhang and Studer [6] published a review on the use of isonitriles as radical acceptors.
There are some examples of imidoyl radicals obtained from amides: Esker and Newcomb [7] prepared imidoyl radicals from secondary amides by reaction of the amide with phosgene to give an imidoyl chloride followed by reaction with the sodium salt of N-hydroxypyridine-2-thione which produced amidyl radicals by visible light initiation. These radicals were cyclized to form γ-lactams and N-acyl pyrimidines. Manley [8] and Zhong et al. [9] obtained imidoyl chlorides by the combination of oxalyl chloride and DMF, and Cunico and Pandey [10] obtained these compounds from amides and phosphorous pentachloride.
The transformation of imidoyl chlorides to selenoimidates or teluroimidates facilitates the handling of the imidoyl radicals; these compounds are prepared from imidoyl chlorides and phenylselenyl anions [11] or from the reaction of oxime sulfonates with organoaluminum selenoates [12] . Bowman et al. [13] obtained phenyl-selenyl imidates from the reaction of aromatic ortho-alkenylamides with phosgene and phenyl selenide and Fujiwara et al. [11] made indoles from butyl-selenyl imidates prepared from selenium and isocyanates.
Bachi and Dennenmark published a short paper [14] on the cyclization of selenoimidates to obtain chromanones or chromenoquinolines from 2-allyloxy-N- alkylbenzamides or 2-allyloxy-N-phenylbenzamides as raw materials; they report imidoyl radicals as intermediates of the cyclization reactions, but the methodology for radical preparation is not reported. After these papers, they did not use this method. Rigby et al. [15] and Fujiwara et al. [11] also prepared and characterized some selenoimidates.
A recent review [16] reports various radical cyclizations of isocyanides, isothiocyanates, nitriles, and other C-N unsaturated bond systems. A variety of 5- and 6-membered nitrogen-containing heterocycles were prepared via imidoyl or iminyl radical intermediates.
2. Results and Discussion
We prepared 2(2-alkenyloxy)-alkyl-selenoimidates from methyl salicylate and butyl amine to obtain the correspondent amides which were made to react with prop-2-enyl bromide or but-2-enyl bromide. The imidoyl chlorides were prepared from 2-(alk-2-en-yloxy)-N-butyl benzamides and oxalyl chloride in anhydrous dichloromethane at −10˚C, according to Booshard [17] . Further reaction with sodium phenyl-selenide as reported by Dowd [18] produced N-butyl- imidoyl selenides (1).
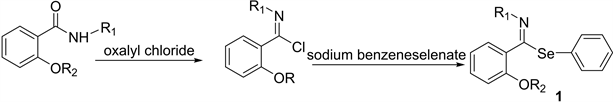
These unstable compounds, solids at room temperature, were purified after basification using chromatography on basic alumina. Yields of 79% - 75% were obtained. Their IR spectra and H1NMR spectra were the expected, as reported by Meese et al. [19] .
2(N-alken-2-enylamino)-N-butyl benzamides (2) were prepared from the reaction of isatoic anhydride with sodium hydride in anhydrous DMF. This anion reacted either with allyl or crotyl amines and finally with butylamine:

2-(N,N-dialkenylamino)-N-phenylbenzamides were prepared from the allylation of isatoic anhydride with sodium hydride and alkenyl bromide in DMF followed for excess of aniline to obtain 2,2-N-alkenyl-N-phenylbenzamides. The resulting N-allyl isatoic acid reacted with alkenyl bromide in DMF(3).
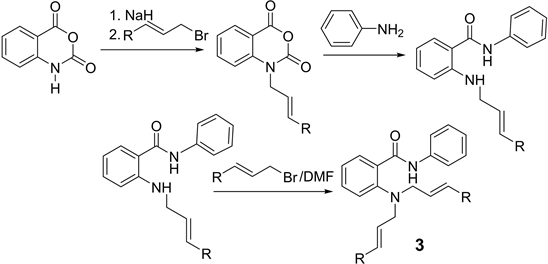
Synthesis of 2-(N,N-dialk-2-enylamino)-N-phenylbenzamides
The phenylamide of 2-aminonicotinic acid (1i in Table 1), was prepared from 2-aminonicotinic acid through the formation of an oxathiazinone with thionyl chloride [20] ; further treatment with sodium hydride in DMF-allyl bromide and finally with aniline. Extraction and chromatography produced 2-(N-allylamino)- N-phenyl nicotinamide. This compound was methylated with sodium hydride and methyl bromide to give 2(allyl(methyl)amino-N-phenyl nicotinamide
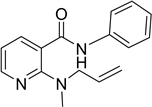
2-(allyl(methyl)amino-N-phenyl nicotinamide
Sodium phenyl selenides were prepared from diphenyl diselenide and sodium hydride in dry THF under dry nitrogen, an added to the imidoyl chlorides in the presence of dimethylformamide to get selenoimidates [21] . Yields of these reactions (60% and 70%) were comparatively higher than the reported (20% - 45%) (see Table 1). We did not find nitriles produced by fragmentation. In all cases, a small quantity of amide was recovered (see Table 1). We did not find nitriles produced by fragmentation recovered.
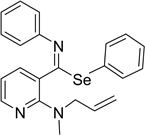
Phenyl (E)-2-allyl(methyl)amino)-N-phenylpyridine-3-carbimidoselenoate 1i

Table 1. Yields of allyl or crotyl amides and selenoamides and selenoimidataees.
・ products of 2-aminonicotinic acid.
Two examples of the NMR 1H spectra of benzamides and two of the imidoyl chlorides are shown:
Benzamides

2-(propen-yloxy-N-phnyl) benzamide 1HRMN
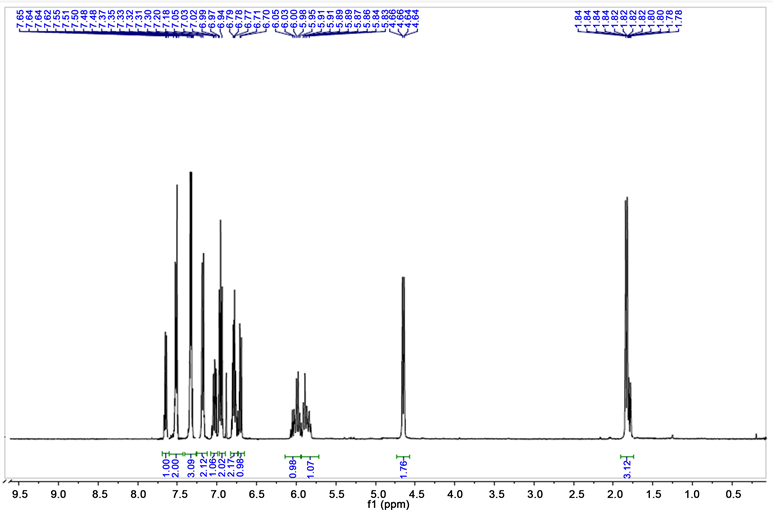
2(N,N-dibut-2-enediamino)-N-benzamide
Imidooyl selenoimidates:
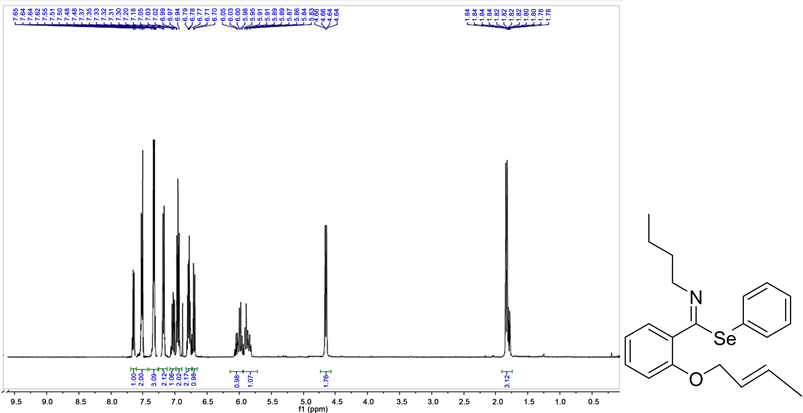
Phenyl(E)-2-(((E)-but-2-en1-yl)oxy)-N.butyl-benzimidoselenoate
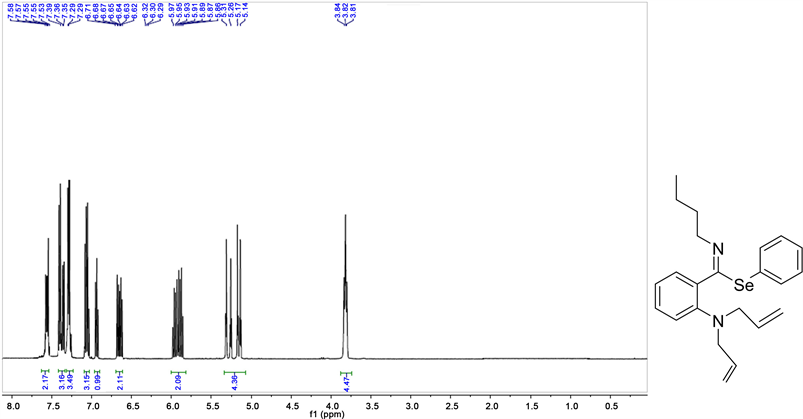
Phenyl ((E)-2-diallylamino)-N-phenylbenzimidoselenoate
Free radical cyclization.
The cyclization of phenyl-N-alkyl benzimidoselenoates to obtain 4-cromanones and 4-quinolones (2) was carried out in free radical conditions through the slow addition of 1 equivalent of tri-N-butyltin hydride and 0.25 eq. of AIBN in dry toluene [16] [17] . The imidoyl radical was trapped by the double bond to produce heterocycles fussed to the aromatic ring: 3-Metyl-2,3-dihidro-4-cromanone (73%) and 3-ethyl-2,3-dihidro-4-cromanone (64%) (2, X=O) were obtained after the hydrolysis of the resulting. exocyclic imine from 2-(2-alkenyloxi)-ben- zamidoyl imidates and 2,3-dihidro-3-methyl-N-prop-2-enyl-4-quinolone 2 (X=N) (82%) and 2,3-dihidro-3-ethyl-N-but-2-enyl quinolones (64%) were obtained from phenylselenyl-(N,N-dialkenylamine)-N-butylbenzoimidates. This method had not been reported for the synthesis of quinolones.
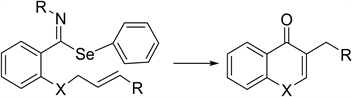 X = O or NR.
X = O or NR.
Examples of the NMR 1H spectra of chromanones and quinolones are shown:
Tandem cyclization
Applying the previous experimental procedure to N-phenyl substituted imidoylselenoates, we obtained only one cyclization, therefore we decided to duplicate AIBN and tributyltin hydride addition times. This modification produced a cascade cyclization. In early experiments, we isolate four products whose structures were assigned from chromatography-mass spectra, IR and NMR analysis. Besides the main product of molecular weigh 261 (4), reported by Bachi and Denenmark [10] , with an intense fluorescence in UV light. Two slightly fluorescent products with molecular weight 263 were identified (5), presenting a

3-ethyl-2,3-ldihydro-4-chromanone 1HRMN
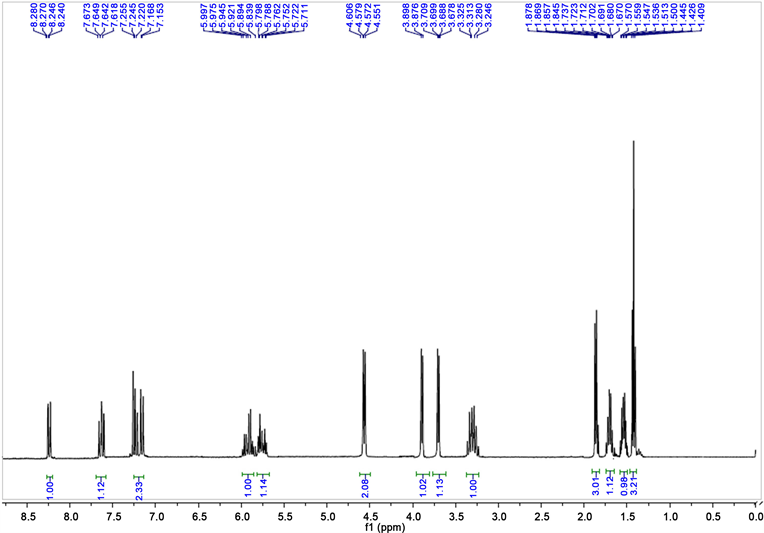
2,3-dihydro-(N-but-2-enyl)4-quinolone 1HNMR
N-H band at 1464, the stretching C=N and the stretching N-H band 3247 in de IR spectra. IR signals were assigned to the enamine-imine equilibrium of the direct free radical addition product. Two more compounds of molecular weight 269 were isolated and identified (5) which are, probably, hydrolysis products of the imino-enamine mixture.

Chromenoquinolines
Method optimization: the slow addition of two hydride equivalents of tri-n-butyltin hydride and 0.03 equivalents of AIBN, produced the radical (6-exo-trig) cyclization with the double bond, to generate a new radical which reacted with the benzene ring. The addition of DDQ in toluene produced the cromenoquinoline. (6)

The obtained yields of 6H-chromeno [4,3,b] quinoline (41%) from the and of 7-methyl-6H-chromenoquinoline (67%) can be explained considering the stability of the primary or secondary intermediate radicals.
Dibenzonaphtyridines
The cyclization of 2,2-alkenylamino-N-phenyl-benzamides through the previous methodology produced 5,6-dihydro-dibenzo [b,h][1,6] naphthyridine in 49% yield and 5,6-dihydro-7-methyl-dibenzo [b,h][1,6] naphthyridine in 71% yield (7). We also carried out the cyclization of 2-(N,N-di-prop-2-enylamino)- N-phenyl-nicotinamide selenoate obtained from 2-aminonicotinic acid by adding the sodium hydride an de radical initiator during 5 hours. The 5,6-dihidro- benzo[b]pyrido2,3-h][1,6] naphthyridine (8) was obtained in 59% yield.
 R=H, CH3 7
R=H, CH3 7  8
8
NMR 1H spectra of 7-methyl-6H-chromeno[4,3-b]chromenoquinoline and of 5,6-dihydro[-7-methyl-dibenzo(b,h)[1,6] naphthyridine are shown:

7lmehyl-6H-chromeno(3,4)chromenoquinoline

5,6-dihydro[7-methyl-dibenzo(b,h)]1,6,naphthiridine
This cyclization method was assessed for the cyclization of the aromatic Imidoyl chlorides, ra5,6w materials of the imidoyl chlorides, which are stable enough, but the yields were very low.
3. Experimental
The raw materials and silica gel were acquired from Sigma Aldrich. DMF was dried with calcium hydride and dichloromethane with phosphorous pentoxide. Toluene and THF were distilled over Na/benzophenone prior used. Melting point was determined with a Fisher-Johns apparatus. HNMR spectra were measured on a Varian Unity Inova 300 MHz using CDCl3 as solvent. Chemical shifts are reported in ppm downfield from tetramethyl-silane. Data are presented as follows: Chemical shift, multiplicity (s: singlet; d: doublet: t: triplet, m: multiplet), coupling constant in hertz and integration. Mass spectra measurements were performed on a Thermo-Electron DFS spectrometer coupled to a gas chromatographer, Thermo-Electron GC Ultra. UV spectroscopy was obtained in a Lamda-2-Perkin Elmer spectrometer.
N-butylsalicylamide: From 2.5 g (16.44 mmol) of methyl salicylate and 3.9 ml (40 mmol) of butylamine. 6 g (86%). Liquid purified by chromatography (hexane, AcOEt 80:20) IR (cm−1): 3399, 2929, 1649, 1299, 1233, 756, 1HNMR: 1.9 ((t,3H), 1.4 - 1.6 (m, 4H) 3.8 (q, 2H), 6.8, (d, J = 8.9 Hz, 1H), 7.0 (d, J = 9.1 Hz), 7.2, (s, 1H) 7.25 (d, J = 8.9 Hz, 1H), J = 8.5 Hz J = 0.9 Hz), 7.6 (t, J = 8.9, 9.1 Hz, 1 H), 11 (s, 1H).
N-phenylsalicylamide: Ethyl-magnesium bromide (two equivalents) in anhydrous THF was added to aniline (two equivalents) with vigorous stirring. The resulting anion was added to methyl-salicylate (one equivalent) and heated for three hours under reflux. After neutralization, the amide was extracted with ethyl acetate. Solvent evaporation and recrystallization produced N-phenyl-salicylamide. M.p. 134-5, IR (cm−1): 3407, 3060, 1633, 1597, 1454,749. 1HNMR (300 MHz, CDCl3) δ (ppm): 6.8 - 7.1 (m 4H), 7.2 - 7.6 8(m.5H), 8.0 (dd, 1H J 0.9 J = 8.5 Hz).
2-Alkenyloxy-N-alkylbenzamides: Allyl or crotyl bromide were added to N-alkyl and N-aryl salicylamides in acetone and refluxed for 10 hours. Elimination of the inorganic salts and solvent evaporation produced the alkenyloxi-N-alkylbenzamide which was purified by column chromatography.
2-(but-2-enyloxy)-N-butylbenzamide: From 2.5 g (12.95 mmol of salicylamide and 1.5 ml crotyl bromide). 2.9 g (89.2%) from column chromatography. IR (cm−1): 3399, 2956, 1652, 1481, 1227, 1102, 755. 1HNMR: (300 MHz, CDCl3) d (ppm): 0.9 (t, 3H) 1.4 (m, 2H), 1.6 (m, 2H), 1.78 (dd J = 1.8, J = 6.6 Hz 3H), 4.5 (m,2H), 5.7 (m, 1H), 5.9 (m, 1H, J = 1 Hz), 6.9 (dd, J = 1 Hz, 1H), 7.3 (td, J = 8.5, 0.9 Hz 1H), 7.9 (s, 1H) 8.2 (d J = 7.5 1H).
2-(prop-2.-enyloxy)-N-butylbenzamide. From 1.47 g (765 mmol) of salicylamide and 1.0 ml 11.48 mmol of allyl bromide. 79% (prop-2-enyloxy)-N-butylbenzamide. m.p. 38 - 40 m.p.38˚C - 41˚C. IR (cm-1): 3355, 2955, 2923, 1666, 1597, 1540, 1317, 751. NMR (300MHz,CDCl3) (ppm): 0.93 (t, 3H, J = 7.2 Hz), 1.33 - 1.45 (m, 2H9 1.5 - 1.61 m, 2H, 3.41 - 3.47 m, 2H) 4.64 (dt, 2H, J = 5.7, J = 1.5 Hz) 6.01 - 6.14 (m, 1H), 6.93 (dd, 1H J = 8.4 Hz, 0.6 Hz), 7.01 - 7.11 (m, 1H), 7.36 - 7.42 (m, 1H), 7.93 (s, 1H), 8.30 (dd, 1H, J = 7.81 Hz J = 2.1 Hz).
2-(prop-2-enyloxy)-N-phenylbenzamide: From the raction of N-phenylbenzamide (2.45 g) (11.5 mmol), 1.5 ml (17.32 mmol) of allyl bromide and 1g of potassium carbonate. (Yield 87%) m.p. 46 ˚C - 49 ˚C. IR (cm−1): 3328, 1660, 1596, 1545, 1318, 1224, 748. HNMR (300 MHz, CDCl3) δ (ppm): 4.73 (d, 2H, J = 6 Hz), 5.46 (dd, 1H, J = 10.5 Hz, 1.2 Hz), 5.54 (dt, 1H, J = 17.25 Hz, 1.5 Hz), 6.10 - 6.25 (m, 1H), 7.0 (d, 1H, J = 9 Hz), 7.08 - 7.15 (m, 2H), 7.35 (t, 2H J = 8.4 Hz), 7.43 - 7.49 (m, 1H), 7.66 (dd, 2H, J = 8.55 Hz, 1.2 Hz), 8.30 (dd, 1H, J = 7.95 Hz 2.1 Hz), 9.9 (s, 1H).
2-(N-alken-2-enylamino)-N-phenyl benzamides:
Isatoic anhydride (1 g, 6.12 mmol) in anhydrous DMF was made to react with sodium hydride for 1 hour, followed of de addition of allyl or crotyl bromides and heating for 1 hour. The product was poured into aniline 5 ml (64 mmol) and stirred for 1 hour. The organic layer was extracted with ethyl acetate and the aniline excess was distilled under reduced pressure. The product was purified by silica gel column chromatography with hexane-ethyl acetate 80:20.
2-(N-prop-2-enylamino)-N-phenyl benzamide: From 1.0 g. isatoic anhydride (6.12 mmol), 0.8 ml allyl bromide (9 mmol) (89%). M.p. 97˚C - 99˚C, IR (cm−1): 3340, 3312, 1655, 1598, 1550, 1224, 1001, 744.
2-(N-but-2-enylamino)-N-phenyl benzamide: From 1.0 g isatoic anhydride (6.12 mmol), 0.95 ml allyl bromide (9 mmol) (81%). M.p. 97˚C - 99˚C IR (cm−1): 3341, 3314, 1654, 1597, 1547, 1478, 1223, 744.
2-N,N-(dialkenylamino)-N-phenyl benzamides.
2-(N-alk-2-enylamino)-N-phenyl benzamides (1.5) g in DMF (60 ml), potassium carbonate (0.9 g) and allyl or crotyl bromide (1.5) eq. were refluxed 7 h. The solvent eliminated at reduced pressure and the product chromatographed in a silica gel column, using hexane-ethyl acetate 80:20.
2-N,N-(di-prop-2-enylamino)-N-phenylbenzamide: From allyl bromide (0.8 ml, 8.9 mmol). Pale yellow crystals. Yield 89%. M. p. 56˚C - 59˚C. IR (cm−1): 3342, 1657, 1597, 1548, 1001, 744, 748. NMR (300 MHz, CDCl3) δ (ppm): 3.83, (t, 4H, J = 5.4 Hz), 5.1 (dc (2H, J = 10.5, 1.5 Hz), 5.29 (dc, 2H, J = 10.5, 1.5 Hz), 5.29 (dc, 2H, J = 16.95, 1.8 Hz), 5.84 - 5.98 (m. 2H). 6.60 - 6.72 (m, 2H), 7.14 (t, 1H, J = 7.5 Hz, 1.5 Hz), 7.29 - 7.37 (m, 3H), 7.48 (dd 1H, J = 7.9, 1.5 Hz)), 7.47 - 7.58 m, 2H), 7.76 (s, 1H).
2-N,N-(di-buten-2-ylamino)-N-phenylbenzamide. From crotyl bromide (1.0 ml, 9.62 mmol). Pale yellow crystals. Yield 87%. M. p. 61˚C - 62˚C. IR (cm−1): 3341, 1655, 1597, 1548, 1478, 1223, 744, NMR (300 MHz, CDCl3) δ (ppm): 1.61 (dd, 6H, J = 9 Hz, 2.5 Hz), 3.80 - 3.84 (m, 4H), 5.85 - 5.97 (m, 2H), 5.92 - 6.15 (m, 2H), 6.59 - 6.71 (m, 2H), 7.13 (tt, 1H, J = 7.5 Hz, 1.8 Hz), 7.27 - 7.39 (m, 3H), 7.47 (dd. 2H, J = 7.8, 1.5 Hz), 7.51 - 7.60 (m, 2H), 7.55 (s, 1H).
4(1H)-oxo-pyrido(2,3-d) oxathiazinone
1.8 ml (21.7 mmol) of thionyl chloride were slowly added to a CCl4 solution of 2-aminopyridine-carboxylic acid (2 g. 14.49 mmol) at 0˚C, After stirring 4 h, 4 (1H)-oxo-pyrido(2,3-d) oxathiazinone was obtained as an orange solid m.p. 171˚C - 173˚C, NMR (ppm): 6.88 (t, 1H, J = 6.94 Hz), 7.35 (s, 1H), 8.0 (dd 1H, J = 6.3, 1.5 Hz), 8.59 (dd 1H, J = 7.5, 1.5 Hz).
2-(N,N-di-prop-2-enylamine)-N-phenylnicotinamide: 4(1H)?oxo-pyrido-(2,3-d) oxathiazinone (1.5 g) in anhydrous DMF were made to react with 0.3 g (12.5 mmol of sodium hydride stirring for 1 hour. 5 ml. (12.2 mmol) or allyl bromide were added to the anion and the mixture refluxed during 12 hs and poured in a suspension aniline (6 ml, 65.8 mmol)-water at 0˚C. After stirring 2 hours and maintained at 0˚C for 10 hours. The organic layer was separated from water and the aniline distilled at reduced pressure. The resulting product was purified by chromatography on a silica gel column.
The 2-N-prop-2-enylamino)-N-phenyl-nicotinamide (54%) is a pale yellow solid, m.p.103˚C - 108˚C. IR: 3354, 3328, 1668, 1593, 1538, 750. This compound and 1.2 g of potassium carbonate and 0.6 ml of allyl bromide in 60 ml of DMF was refluxed for 7 hours. After isolation, the product was purified by column chromatography using hexane-ethyl acetate as eluent. A yellow solid was obtained. Yield: 79%. M. p: 113˚C - 116˚C. IR (cm−1): 3354, 1665, 1596, 1538, 1451, 1317, 750, 1HNMR. δ (ppm): 3.81 (t, 4H, J = 5.4 Hz), 5.16 (dc, 2H, J = 16.95 Hz 1.8 Hz), 5.27 (dc, 2H, J = 16.95 Hz, 1.8 HZ) 5.27 - 7.39 (m, 3H), 7.47 (dd, 1H, J = 8.2, 1.5 Hz), 7.53 (dt, 2H, J = 7.5 Hz, 0.9 Hz) 7.77 (s, 1H), 8.13 (dd, 1H, J = 8.4 Hz, 0.6 Hz).
1,6-dihydro-benzo-[b]-pyrido-[2,3,h]-naphthyridine (Yield 58%) IR: 3307, 1627, 1615, NMR (300 MHz, CDCl3) δ (ppm): 5.15 (s,1H), 5.37 (d, 2H, J = 6.5 Hz), 7.14 (1 H, td, J = 6.0, 2.3 Hz), 7.67 (td 1H J = 9, 2.5 Hz), 7.72 - 7.89 (m, 2H), 8.07 (dd, 1H, J = 9.2 Hz), 8.20 ppm (td, 1H, J = 8, 1.5 Hz), 8.32 (1H dd, J = 8.8, 1.5 Hz).
Imidoyl radicals cyclization.
In a two necked flask fit with a condenser and nitrogen atmosphere, a 0.02 M solution of phenyl selenium imidate or imidoyl chloride (1 eq) in anhydrous toluene was bubbled with nitrogen in order to eliminate the oxygen. 1 eq. of tributiltin, and 0.025 eq of AIBN in toluene (15 ml) were placed in a sealed vial, under nitrogen. Theimidate solution was heated at 100˚C and the mixture of Bu3SnH-AIBN was slowly added during 2 hours. The mixture was refluxed 2 more hours. After cooling at room temperature, 20 ml of a 4 M aqueous solution of sodium hydroxide was added. After stirring 2 hrs, the organic layer was washed with water until pH 7, and dried with sodium sulfate. The residue of evaporation of the solvent at reduced presion was purified in a silica gel column, using hexane-ethyl acetate 93:7 as eluent.
3-methyl 2,3-dihydro-4-chromanone.
From 0.4 g (1.07 mmol) of Phenyl-2-(N,N-diprop-2-enyloxy) N-phenyl benzimidoselenoate. (Numero) (1.07 mmol). Orangeoil. 73% yield. IR (cm−1): 2950, 2851, 1692, 1606, 1296, 1015, 754. 1NM (300 MHz, CDCl3), (ppm): 1.37 (dd, 3H, J = 6.0, 0.9 Hz), 3.41 (m.1H), 4.18 (dm 1H, J = 10 Hz), 4.34 (d, 1H, J = 11.5 Hz), 6.66 (dd, 1H, J = 8.4, 0.6 Hz), 6.71 - 6.77 (m, 1H), 7.01 - 7.08 (m 1H), 7.69 (dd, 1H, J = 7.8, 2.1 Hz). EM (m/z) M+ 162 (70%), 120 (45%), 92 (100%), 77 (10%), 64 (40%).
3-ethyl-2,3-dihydro-4-chromanone.
From 0.4 g (1.03 mmol of phenyl-2-(N,N-dibut-2-enyloxy) N-butyl benzimidoselenoate). Orange liquid. (64%). IR (cm−1): 2956, 2923, 1699, 1296, 754. 1NMR (300 MHz, CDCl3), (ppm): 0.93 (t, 3H J = 9 Hz), 3.17 - 3.41 (m.1H), 4.33 (dt 1H, J = 9, 1.5 Hz), 4.49 (d, 1H, J = 10.5 Hz), 6.78 (dd, 1H, J = 8.4, 0.6 Hz), 6.84 - 6.91 (m, 1H), 7.15 - 7.23 (m 1H), 7.79 (dd, 1H, J = 7.81, 2.1 Hz). EM (m/z) M+ 176 (80%), 120 (50%), 92 (100%), 77 (15%), 64 (40%).
2,3-dihydro-3-methyl-N-prop-2-enyl-4-quinolone.
From 0.45 g (1.09 mmol of phenyl-2-(N,N-dibut-2-enyloxy) N-butyl benzimidoselenoate). Orange liquid (82%). IR (cm−1): 2956, 2925, 2872, 2855, 1739, 1596, 1545, 1440, 1318, 1223, 748. 1NMR (300 MHz, CDCl3), (ppm): 1.47 (dd, 3H, J = 6.5, 1.2 Hz), 3.14 - 3.19 (m.1H), 4.0 (dm 1H, J = 9.5 Hz), 4.15 (d, 1H, J = 10.5 Hz), 4.75 - 4.79 (m, 2H), 5.20 (dd, 1H J = 10.25, 0.9 Hz), 7.22 - 7.27 (m.1H), 7.63 (dt, 1H, J = 7.65, 1.5 Hz), 8.23 (dd, 1H, J = 7.8, 1.8 Hz). EM (m/z): M+ 201, (60%), 161 (45%), 91 (10%), 77 (10%), 64 (40%).
Cyclization of imidoyl radicals to obtain cromenoquinolines and dibenzonaphtyridines.
In a two necked flask fit with a condenser and nitrogen atmosphere, a 0.02 M solution of phenyl selenium imidate or imidoyl chloride (1 eq.) in anhydrous toluene was bubbled with nitrogen to eliminate oxygen. 2 eq. of tributyltin hydride, and 0.03 eq. of AIBN in toluene (15ml) were placed in a sealed vial, under nitrogen. The solution was heated at 100oC and a mixture of Bu3SnH-AIBN was slowly added during 4 hours and refluxed 2 hours. To the resulting solution 1.1 eq. of DDQ, was added and heated 1.5 hours. After cooling, the solution was stirred 2 hours with 30 ml or a 4 M solution of NaOH. The organic layer washed with water until pH 7 was dried over sodium sulfate. The residue of the solvent evaporation at reduced pressure was purified in a silica gel column, using hexane-ethyl acetate 93:7 as eluent.
6H-cromeno [4,3-b] quinolone 9a.
From 0.6 g (1.53 mmol) of Phenyl-2-(prop-2-enyloxy) N-phenyl benzimidoselenoate. Yield: 41%. Orange solid. M.p. 112˚C - 113˚C, IR (cm−1): 3062, 2959, 2926, 2853, 1651, 1597, 1522, 1438, 1307, 1248, 757. 1NMR (300 MHz, CDCl3), (ppm): 5.30 s, 2H, 7.00, (dd, 1H, J = 7.5, 1.2 Hz), 7.13 (td, 1H, J = 8, 1n5 Hz), 7.32 (td, 1H, J = 7.5, 1.5 Hz), 7.47 (td, 1H, J = 6.7, 0.9 Hz), 7.62 - 7.73 (m, 2H) 7.84 (s, 1H), 8.707 (dd, 1H, J = 8, 1.7 Hz) 8.45 (dd 1H, J = 8, 1.5 Hz). EM (m/z): M+ 233 [M+], (62%), 142 (100%), 103 (10%), 92 (80%), 77 (20%).
7-methyl-6H-cromeno [4, 3-b] quinoline.
From 0.66 g (1.59 mmol) Phenyl-2-(N-but-2-enyloxy) N-phenyl benzimidoselenoate. Yield: 67%. Orange solid. M.p. 121˚C - 123˚C, IR (cm−1): 3060, 2924, 2852, 1651, 1597, 1522, 1438, 1307, 1248, 757. 1NMR (300 MHz, CDCl3), (ppm): 5.11 (s, 1H), 5.30 (d, J = 6 Hz, 7.0 (dd, 1H, J = 7.5, 1.3 Hz), 7.33 (td, 1H, J = 7.5, 1.5 Hz), Hz), 7.49 (s, 1H), 7.73 (td, 1H, J = 7.5, 0.9 Hz), 7.77 - 7.86 (m, 2H), 8.07 (dd, 1H, J = 7.5, 1.57 Hz), 8.32 (dd 1H, J = 8.0, 0.95. EM (m/z): 232 [M+], (63%), 155 (100%), 141 (100%) 91 (81%), 77 (25%).
5,6-dihydro-dibenzo [b, h] [1,6] naphtyridine.
From 0.7 g (1.59 mmol) Phenyl-2-(N,N-dibut-2-enyloxy) N-phenyl benzimidoselenoate. Yield: 71%. Red solid. M.p. 101˚C - 104˚C, IR (cm−1): 3416, 2957, 2872, 1647, 1599, 1530, 1295, 902, 752. 1NMR (300 MHz, CDCl3), (ppm): 5.16 (s, 1H), 5.37 (d, 2H, J = 6.5 Hz), 7.15 (td, 1H, J = 6,0 2.3 Hz), 7.47 (s, 1H), 7.67 (td, 1H, J = 9.00, 2.5 Hz), 7.72 - 7.89 (m, 2H), 8.07 (dd, 1H, J = 9.0, 2.0 Hz), 8.2, (td 1H, J = 8.0, 1.5 Hz). EM (m/z): 233 [M+], (50%), 141 (100%), 92 (78%), 67 (30%).
Conflicts of Interest
The authors declare no conflicts of interest regarding the publication of this paper.
Cite this paper
Vizcaíno-Escobedo, K.C., Sánchez-Carrillo, J.A. and Albores-Velasco, M. (2018) Imidoyl Radicals for the Synthesis of Chromenoquinolines and Naphthyridines. International Journal of Organic Chemistry, 8, 319-334. https://doi.org/10.4236/ijoc.2018.83024
References
- 1. Curran, D.P. and Liu, H. (1991) 4 + 1 Radical Annulations with Isonitriles: “A Simple Route to Cyclopenta-Fused Quinolines”. Journal of the American Chemical Society, 113, 2127-2128. https://doi.org/10.1021/ja00006a033
- 2. Nanni, D., Pareeschi, P., Rizzoli, C., Sgaraboto, P. and Tundo, A. (1995) Radical Annulation and Ciclization with Isonitriles: The Fate of the Intermediates Imidoyl and Cyclohexadienyl Radicals. Tetrahedron, 51, 9045-9062. https://doi.org/10.1016/0040-4020(95)00348-C
- 3. Nanni, D., Pareschi, P. and Tundo, A. (1996) Isonitriles as Source and Fate of Imidoyl Radicals: A Novel Homolytic-Fragmentation. Tetrahedron Letters, 37, 9337-9340. https://doi.org/10.1016/S0040-4039(97)82958-9
- 4. Zhang, B. and Studer, A. (2015) Recent Advances in the Synthesis of Nitrogen Heterocycles via Radical Cascade Reactions Using Isonitriles as Radical Acceptors. Chemical Society Reviews, 44, 3505-3521. https://doi.org/10.1039/C5CS00083A
- 5. Leardini, R., Nanni, D. and Zanardi, G. (2000) Radical Addition to Isonitriles: A Route to Polyfunctionalized Alkenes through a Novel Three Component Radical Cascade Reaction. Journal of the American Chemical Society, 65, 2763-2772.
- 6. Danoh, Y., Matta, H., Uemura, J., Watanabe, H. and Uneyama, K. (1995) Generation and Reactions of Trifluoroaceimidoyl Radicals. Bulletin of the Chemical Society of Japan, 68, 1497-1507. https://doi.org/10.1246/bcsj.68.1497
- 7. Esker, L. and Newcomb, M. (1993) Chemistry of Amidyl Radicals Produced from N-Hydroxypyridine-2-thione Imidate Esters. The Journal of Organic Chemistry, 58, 4933-4940. https://doi.org/10.1021/jo00070a033
- 8. Manley, P.J. and Bilodeau, M.T. (2002) A Mild Method for the Formation and in Situ Reaction of Imidoyl Chlorides: Conversion of Pyridine-1-Oxides to 2-Aminopyridine Amides. Organic Letters, 4, 3127-3129. https://doi.org/10.1021/ol0264556
- 9. Zhong, Y.L., Kee, J., Reamer, R.A. and Askin, D. (2004) New Method for the Synthesis of Diversely Functionalized Imidazoles from N-Acylated α-Aminonitriles Organic Letters, 6, 929-631. https://doi.org/10.1021/ol036423y
- 10. Cunico, R.F. and Pandey, R.K. (2005) Palladium Catalyzed Synthesis of a Iminoamides from Imidoyl Chlorides and a Carbamoyl Silane. The Journal of Organic Chemistry, 70, 5344-5346. https://doi.org/10.1021/jo050545h
- 11. Fujiwara, S., Maeda, H., Matsuya, T., Shin-Ike, T., Kambe, N. and Sonoda, N. (2000) Imidoylation of Acidic Hydrocarbons with Selenium and Isocyanides: A New Synthetic Method for Preparation of Sele-noimidates. The Journal of Organic Chemistry, 65, 5022-5025. https://doi.org/10.1021/jo0001479
- 12. Maruoka, K., Miyazaki, T., Ando, M., Matsumura, Y., Sakane, S., Hattori, K. and Yamamoto, H. (1983) Organoaluminum-Promoted Beckmann Rearrangement of Oxime Sulfonates. Journal of the American Chemical Society, 105, 2841-2843. https://doi.org/10.1021/ja00347a052
- 13. Bowman, W.R., Fletcher, A.J., Pederson, J.M., Lovell, P.J., Elsegood, M.R.J., Lopez, E.H., McKee, V. and Potts, G.B.S. (2007) Amides as Precursors of Imidoyl Radicals in Cyclization Reactions. Tetrahedron, 63, 191-205. https://doi.org/10.1016/j.tet.2006.10.030
- 14. Bachi, M.D. and Denenmark, D. (1989) Cyclizations of Ene Radicals. Imidoyl Radicals as Intermediates in the Synthesis of Heterocyclic Compounds. Journal of the American Chemical Society, 111, 1886-1888. https://doi.org/10.1021/ja00187a057
- 15. Rigby, J.H., Danca, D.M. and Horner, J.H. (1998) Carbamoyl Radicals from Phenylselenocarbamates: Intramolecular Additions to Alkenes. Tetrahedron Letters, 39, 8413-8416. https://doi.org/10.1016/S0040-4039(98)01830-9
- 16. Lei, J., Li, D. and Zhu, Q. (2018) Synthesis of Nitrogen Containing Heterocycles via Imidoyl or Iminyl Radical Intermediates. Topics in Heterocyclic Chemistry, 54, 285-320.
- 17. Bosshard, H.H., Mory, R., Schmid, M. and Zollinger, H. (1959) Eine Methode sur katalysierten herstellung von Carbonsaure und Sul-fosaure-chloriden mit Thionylchlorid. Helvetica Chimica Acta, 42, 1653-1658. https://doi.org/10.1002/hlca.19590420526
- 18. Dowd, P. and Kennedy, P. (1981) Convenient Preparation of Sodium and Potassium Phenyl Selenides. Reduction of Selenium-Selenium Bond by Sodium Hydride and Potassium Hydride. Synthetic Communications, 11, 935-941. https://doi.org/10.1080/00397918108065751
- 19. Meese, C.O., Walter, W., Mrotzek, H. and Mirzaí, H. (1976) Selenoimidsaureester. Chemische Berichte, 109, 956-964. https://doi.org/10.1002/cber.19761090316
- 20. Zografos, A. and Mitsos, C. (2001) Chemoselective Cyclization of Aminonicotinic Acid Derivatives to 1,8-Naphthyridin-2-ones via a Potential Intramolecular Azadiene-Ketene Electrocyclization. The Journal of Organic Chemistry, 66, 4413-4415. https://doi.org/10.1021/jo0057553
- 21. Crich, D. and Fortt, S.M. (1987) On the Effect of Ring of Ring Size in the Cyclization of Carbonyl and Vinyl Radicals onto Alkenes. Tetrahedron Letters, 28, 2895-2898. https://doi.org/10.1016/S0040-4039(00)96238-5