International Journal of Organic Chemistry
Vol.06 No.01(2016), Article ID:64682,11 pages
10.4236/ijoc.2016.61005
Synthesis and Spectral Identification of Novel Stable Triazene: As Raw Material for the Synthesis Biocompatible Surfactants- Pyrazole-Isoxazole-Dihydropyrimidine- Tetrahydropyridine Derivatives
Mohamed Ahmed Mahmoud Abdel Reheim*, Ahmed Mahmoud El-Sayed Tolba
Department of Chemistry, Faculty of Science, Suez Canal University, Arish, Egypt

Copyright © 2016 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 3 November 2015; accepted 14 March 2016; published 17 March 2016
ABSTRACT
The chemical reactivity of novel stable triazene 3 toward some nucleophilic and electrophilic reagents was investigated. Traizene 3 was used as a key precursor for the synthesis of some novel important heterocyclic compounds such as Pyrazole, Isoxazole, Dihydropyrimidine, Tetrahydropyridine derivatives with expected antimicrobial activity. The synthesized compounds were obtained in good yields. The structures of the newly synthesized compounds were confirmed by elemental analysis, IR, 1H-NMR and Ms spectral data.
Keywords:
Biocompatible Surfactants, Pyrazole, Isoxazole, Dihydropyrimidine Tetrahydropyridine Derivatives, Spectral Characteristics

1. Introduction
Surfactants are organic compounds that have the ability to decrease the surface tension of water and consisting of two moieties: a non-polar hydrophobic group (referred to as the tail) which is usually a straight or branched long hydrocarbon chain which is attached to a polar hydrophilic group (referred to as the head) [1] . Surfactants are widely used in the food, pharmaceutical, cosmetic, textile, paint and coating industries, as emulsifying, wetting and foaming agents, solubilizers and suspension stabilizers [2] [3] . Amino acid-based surfactants are characterized by low toxicity, good biocompatibility and fast biodegradation. Some of these compounds also show antibacterial activity [4] -[8] . Simple nitrogen-containing heterocycles receive a large amount of attention in the literature, as a consequence of their exciting biological properties and their role as pharmacophores of considerable historical importance. Pyrazolones nucleus have attracted much attention due to their interesting biological activities [9] [10] . Pyrazolone derivatives are widely used in medical practice (antipyrine, amidopyrine, analgin, etc.) [11] . A large number of thienopyrimidines were reported in the literature as virucides, bactericides, fungicides, acaricides, insecticides [12] . In this paper, we have reported a variety of synthesizes of heteroaromatics developed using functionally substituted triazene as readily obtainable building blocks possessing multiple electrophilic and nucleophilic moieties of expected biological interest.
2. Material and Methods
2.1. Experimental
All melting points were measured using Akofler Block instrument and are uncorrected. IR spectra (KBr) were recorded on a FTIR 5300 spectrometer (υ, cm−1). The 1H-NMR spectra were recorded in DMSO-d6 and CDCl3 at 300 MHz and 400 MHz on a Varian Gemini NMR. 1000 EX mass spectrometer at 70 ev. The purity of synthesized compounds was checked by thin layer chromatography TLC (aluminum sheets) using n-hexane, ethyl acetate (9:1, V/V) eluent. Elemental analyses were carried out by the Microanalytical Research Center, Faculty of Science, and Microanalytical Unit, Faculty of Pharmacy, Cairo University, Egypt.
2.2. General Procedure for the Preparation of Compounds (3a-c)
The aromatic amines (0.01 mole) were dissolved in (2 M) hydrochloric acid (10 ml), diluted with water (30 ml) and diazotized at 0˚C with sodium nitrite (0.01 mole) in a minimum volume of water. A solution of glycine ethyl ester hydrochloride in water was added slowly to the diazonium salt solution. The mixture was stirred for 1 hr at 0˚C, where upon the solution was normally clear. The clear solution was treated with a large excess of sodium acetate ( 5 gm ) with continuous stirring for 2 hr. The resulting reaction product was filtered off, washed with water and crystallized from the proper solvent to give compounds (3a-c).
1) Ethyl-2-(3-(4-chlorophenyl)triaz-2-enyl)acetate (3a)
Formed as brown crystals from n-hexane; yield (67%); M.P. 60˚C - 62˚C; IR (KBr) n∙cm−1 = 3389 (NH), 3113 (CH-arom), 2974 - 2933 (CH-aliph), 1734 (C=O); 1H-NMR (DMSO-d6) d (ppm) = 1.17 (t, 3H, CH3, J = 3 Hz), 4.13 (q, 2H, CH2-O, J = 6 Hz), 5.02 (br s, 2H, CH2-N), 6.53 - 8.22 (m, 4H, Ar-H), 12.62 (s, 1H, NH); MS: m/z (%) 241 (M+); Anal. Calcd. For C10H12ClN3O2 (241); C, 49.70; H, 5.00; N, 17.39; found C, 49.72; H, 5.01; N, 17.40.
2) Ethyl-2-(3-p-tolyltriaz-2-enyl)acetate (3b)
Formed as brown crystals from n-hexane; yield (71%); M.P. 74˚C - 76˚C; IR (KBr) n∙cm−1 = 3200 (NH), 3020 (CH-arom), 2918 - 2850 (CH-aliph), 1740 (C=O); 1H-NMR (DMSO-d6) d (ppm) = 1.32 (t, 3H, CH3, J = 6 Hz), 1.90 (s, 3H, CH3), 4.10 (q, 2H, CH2-O, J = 6 Hz), 4.42 (br s, 2H, CH2-N), 7.36 - 8.57 (m, 4H, Ar-H), 12.60 (s, 1H, NH); MS: m/z (%) 221 (M+); Anal. Calcd. For C11H15N3O2 (221); C, 59.71; H, 6.83; N; 18.99; found C, 59.70; H, 6.84; N; 19.01.
3) Methyl-4-(3-(2-ethoxy-2-oxoethyl)triaz-1-enyl)benzoate (3c)
The starting compound (3c) was prepared under the same experimental conditions according to a literature procedure [16] .
2.3. 4-(3-(2-Ethoxy-2-Oxoethyl)-Triaz-1-Enyl)-2-Hydroxynaphthalene-1-Sulphonic Acid (3d)
The aromatic amines (0.06 mole) were treated with ( 3.18 gm ) of anhydrous Na2CO3 dissolved in 10 ml of distilled water. The mixture was stirred for 15 min at 40˚C. Where upon the solution was normally clear, cooled mixture at 0˚C. The clear solution was treated with 2.0 gm of NaNO2 dissolved in a minimum volume of water. The solution was cooled in ice water, a mechanical stirrer being used, by the slow addition of (0.2 mole) of HCl from a separatory funnel. A solution of glycine ethyl ester hydrochloride in water was added slowly to the diazonium salt solution. The mixture was stirred for 1 hr at 0˚C. The clear solution was treated with a large excess of NaOH (40 mL, 10%) and the triazene precipitated slowly from the reaction mixture. Precipitation was normally evident after 30 min, and was usually complete within 3 hr. The resulting reaction product was filtered off, washed with water, and crystallized from ethanol to give brown crystals; yield (77%); M.P. 250˚C - 252˚C; IR (KBr) n∙cm−1 = 3400 (NH/OH), 3136 (CH-arom), 2950 - 2850 (CH-aliph), 1700 (C=O); 1H-NMR (DMSO-d6) d (ppm) = 1.05 (t, 3H, CH3, J = 6 Hz), 4.62 (q, 2H, CH2-O, J = 6 Hz), 5.74 (br s, 2H, CH2-N), 7.08 - 9.02 (m, 6H, Ar-H and SO3H), 10.56 (s, 1H, OH), 12.16 (br, 1H, NH); MS: m/z (%) 355 (M++2); Anal. Calcd. For C14H15N3O6S (353); C, 47.59; H, 4.28; N, 11.89; found C, 47.61; H, 4.37; N, 11.90.
2.4. General Procedure for the Preparation of Compounds (5a-c)
A mixture of compounds 3a-c (0.01 mol) and ammonium acetate ( 2 gm ) were fused without solvent for 20 min. The solid precipitate so formed was treated with ethanol and filtered out and crystallized from the proper solvent to give (5a-c).
1) 1-(4-Chlorophenyl)-1H-1,2,3-triazol-5-ol (5a)
Formed as pale yellow crystals from n-hexane; yield (68%); M.P. 70˚C - 72˚C; IR (KBr) n∙cm−1 = 3454 (OH), 3063 (CH-arom); 1H-NMR (DMSO-d6) d (ppm) = 6.93 - 8.06 (m, 5H, Ar-H + CH-triazole), 12.06 (s, 1H, OH); MS: m/z (%) 197 (M++2); Anal. Calcd. For C8H6ClN3O (195); C, 49.12; H, 3.09; N, 21.48; found C, 49.14; H, 3.10; N, 21.47.
2) 1-p-Tolyl-1H-1,2,3-triazol-5-ol (5b)
Formed as pale yellow crystals from n-hexane; yield (68%); M.P. 60˚C - 62˚C; IR (KBr) n∙cm−1 = 3434 (OH), 3060 (CH-arom), 2916 (CH-aliph); 1H-NMR (DMSO-d6) d (ppm) = 1.89 (s, 3H, CH3), 6.70 - 8.49 (m, 5H, Ar-H + CH-triazole), 12.40 (s, 1H, OH); MS: m/z (%) 177 (M++2); Anal. Calcd. For C9H9N3O (175); C, 61.70; H, 5.18; N, 23.99; found C, 61.71; H, 5.19; N, 23.98.
3) Methyl-4-(5-hydroxy-1H-1,2,3-triazol-1-yl)benzoate (5c)
Formed as pale yellow crystals from n-hexane; yield (68%); M.P. 86˚C - 88˚C; IR (KBr) n∙cm−1 = 3448 (OH), 3057 (CH-arom), 2926 (CH-aliph); 1H-NMR (DMSO-d6) d (ppm) = 1.64 (s, 3H, COOCH3), 7.22 - 8.62 (m, 5H, Ar-H + CH-triazole), 12.66 (s, 1H, OH); MS: m/z (%) 219 (M+); Anal. Calcd. For C10H9N3O3 (219); C, 54.79; H, 4.14; N, 19.17; found C, 54.80; H, 4.15; N, 19.16.
2.5. General Procedure for the Synthesis of Palmitoyl Chloride from Palmitic Acid
Palmitoyl chloride was prepared by gently refluxing a mixture of palmitic acid ( 26.6 gm , 0.01 mol) and thionyl chloride (23.7 ml, 0.01 mol) on water bath maintained at 35˚C - 40˚C with shaking. The reaction mixture was refluxed in the water bath for 6 hr. The excess of thionyl chloride was removed by washing successively with water and the residue was dried over anhydrous Na2SO4 to collect the liquid palmitoyl chloride.
2.6. General Procedure for the Preparation of Compounds (7a-d)
A mixture of 3a-d (0.01 mol), and palmitoyl chloride 6 (0.01 mol) in CH2Cl2 (30 ml), pyridine (5 ml) was added to this solution slowly and heated under reflux for 8 hr. The reaction mixture allowed to cool, poured into crushed ice and acidified with HCl. The solid product was filtered off and crystallized from the proper solvent to give (7a-d).
1) Ethyl-2-(3-(4-chlorophenyl)triaz-2-enyl)-3-oxooctadecanoate (7a)
Formed as pale yellow crystals from n-hexane; yield (68%); M.P. 86˚C - 88˚C; IR (KBr) n∙cm−1 = 3310 (NH), 3102 (CH-arom), 2919 - 2850 (CH-aliph), 1659 (C=O); 1H-NMR (DMSO-d6) d (ppm) = 0.84 (t, 3H, CH3, J = 3 Hz), 1.22 - 1.28 (m, 24H, 12CH2), 1.56 (t, 3H, CH3, J = 6 Hz), 2.27 (t, 2H, b-CH2, J = 6 Hz), 2.48 (t, 2H, a-CH2, J = 3 Hz), 2.49 (s, 1H, CH), 3.29 (q, 2H, CH2-O, J = 12 Hz), 7.29 - 7.61 (m, 4H, Ar-H), 9.94 (s, 1H, NH); MS: m/z (%) 479 (M+); Anal. Calcd. For C26H42ClN3O3 (479); C, 65.05; H, 8.82; N, 8.75; found C, 65.07; H, 8.84; N, 8.76.
2) Ethyl-3-oxo-2-(3-p-tolyltriaz-2-enyl)octadecanoate (7b)
Formed as brown crystals from n-hexane; yield (75%); M.P. 63˚C - 65˚C; IR (KBr) n∙cm−1 = 3313 (NH), 3050 (CH-arom), 2953 - 2849 (CH-aliph), 1700, 1661 (2C=O); 1H-NMR (DMSO-d6) d (ppm) = 0.83 (t, 3H, CH3, J = 9 Hz), 1.18 - 1.36 (m, 24H, 12CH2), 1.50 (t, 3H, CH3, J = 6 Hz), 2.13 (s, 3H, CH3), 2.16 (t, 2H, b-CH2, J = 6 HZ), 2.21 (t, 2H, a-CH2, J = 9 HZ), 2.47 (s, 1H, CH), 3.29 ( br s, 2H, CH2-O), 7.04 - 7.45 (m, 4H, Ar-H), 9.69 (s, 1H, NH); MS: m/z (%) 461 (M++2); Anal. Calcd. For C27H45N3O3 (459); C, 70.55; H, 9.87; N, 9.14; found C, 70.58; H, 9.88; N, 9.14.
3) Methyl 4-(3-(1-ethoxy-1,3-dioxooctadecan-2-yl)triaz-1-enyl)benzoate (7c)
Formed as yellow crystals from n-hexane; yield (69%); M.P. 53˚C - 55˚C; IR (KBr) n∙cm−1 = 3459 (NH), 2954 - 2848 (CH-aliph), 1701, 1640 (2C=O); 1H-NMR (CDCl3) d (ppm) = 0.72 (t, 3H, CH3, J = 8 HZ), 1.28 - 1.30 (m, 24H, 12CH2), 1.32 (s, 3H, CH3), 1.63 (t, 3H, CH3, J = 8 HZ), 2.18 (s, 1H, CH), 2.20 (t, 2H, b-CH2, J = 8 HZ), 2.37 (t, 2H, a-CH2 , J = 8 HZ), 3.95 (q, 2H, CH2-O, J = 12 Hz), 6.81 - 8.05 (m, 4H, Ar-H), 11.26 (s, 1H, NH); MS: m/z (%) 503 (M+); Anal. Calcd. For C28H45N3O2 (503); C, 66.77; H, 9.01; N, 8.34; found C, 66.78; H, 9.02; N, 8.35.
4) 4-(3-(1-ethoxy-1,3-dioxooctadecan-2-yl)triaz-1-enyl)-2-hydroxynaphthalene-1-sulphonic acid (7d)
Formed as pale yellow crystals from n-hexane; yield (75%); M.P. 46˚C - 48˚C; IR (KBr) n∙cm−1 = 3462 (NH/OH), 2956 - 2849 (CH-aliph), 1702 (C=O); 1H-NMR (CDCl3) d (ppm) = 0.89 (t, 3H, CH3, J = 8 HZ), 1.27 - 1.30 (m, 24H, 12CH2), 1.32 (t, 3H, CH3, J = 8 HZ), 1.60 (s, 1H, CH), 1.64( t, 2H, b-CH2, J = 8 HZ), 2.35 (t, 2H, a-CH2, J = 8 HZ), 4.30 (q, 2H, CH2, J = 8 Hz), 7.28 - 8.39 (m, 8H, Ar-H + SO3H + OH and NH); MS: m/z (%) 591 (M+); Anal. Calcd. For C30H45N3O7S (591) C, 60.89; H, 7.66; N, 7.10; found C, 60.91; H, 7.65; N, 7.09%.
2.7. Ethyl-2-(3-(4-chlorophenyl)triaz-2-enyl)-3-(dimethylamino)acrylate (8)
A mixture of 3a (0.01 mol) and DMF-DMA (0.01 mol) in dioxane (30 mL) was heated under reflux for 6 hrs. The reaction mixture was allowed to cool. The separated solid was filtered, washed with ethanol and crystallized from ethanol to give brown crystals; yield (78%); M.p.100˚C - 102˚C; IR (KBr) n∙cm−1 = 3378 (NH), 3063 (CH-arom), 2924 - 2853 (CH-aliph), 1667 (CO); 1H-NMR (DMSO-d6) d = 1.33 (t, 3H, CH3, J = 4 Hz) 3.35 (s, 6H, 2CH3), 4.37 (q, 2H, CH2, J = 8 Hz), 7.22 - 8.03 (m, 5H, Ar-H + CH-oliffinic), 12.65 (s, 1H, NH); MS: m/z (%) 296 (M+), Anal. Calcd. For C13H17ClN4O2 (296): C, 52.62; H, 5.77; N, 18.88; Found: C, 52.63; H, 5.78; N, 18.87 %.
2.8. General Procedure for the Preparation of Compound (9a-c)
A mixture of compound 3a (0.01 mol), appropriate aromatic aldehydes (0.01 mol) in ethanol (30 ml) with catalytic amount of piperidine was heated under reflux for 3 hrs. The reaction mixture was allowed to cool and poured into crushed ice then acidified with HCl. The separated solid was filtered, washed with water and crystallized from the proper solvent to give 9a-c.
1) Ethyl 2-(3-(4-chlorophenyl)triaz-2-enyl)-3-phenylacrylate (9a)
Formed as yellow crystals from n-hexane; yield (69%); M.P. 80˚C - 82˚C; IR (KBr) n∙cm−1 = 3387 (NH), 3096 (CH-arom), 2975 (CH-aliph), 1692 (C=O); 1H-NMR (CDCl3) d (ppm) = 1.35 (t, 3H, CH3, J = 3 Hz), 4.37 (q, 2H, CH2, J = 3 Hz), 7.09 - 7.94 (m, 10H, Ar-H + CH-oliffinic), 12.69 (s, 1H, NH); MS: m/z (%) 329 (M+); Anal. Calcd. For C17H16ClN3O2 (329); C, 61.91; H, 4.89; N, 12.74; found C, 61.92; H, 4.90; N, 12.73.
2) Ethyl-2-(3-(4-chlorophenyl)triaz-2-enyl)-3-(4-methoxyphenyl)acrylate (9b)
Formed as yellow crystals from n-hexane; yield (69%); M.P. 60˚C - 62˚C; IR (KBr) n∙cm−1 = 3447 (NH), 3097 (CH-arom), 2932 (CH-aliph), 1676 (C=O); 1H-NMR (DMSO-d6) d (ppm) = 1.22 (t, 3H, CH3, J = 8 Hz), 3.83 (s, 3H, OCH3), 4.44 (q, 2H, CH2, J = 16 Hz), 7.38 - 8.76 (m, 9H, Ar-H + CH-oliffinic), 12.50 (s, 1H, NH); MS: m/z (%) 361 (M++2); Anal. Calcd. For C18H18ClN3O3 (359); C, 60.09; H, 5.04; N, 11.68; found C, 60.10; H, 5.05; N, 11.67.
3) Ethyl-3-(4-chlorophenyl)-2-(3-(4-chlorophenyl)triaz-2-enyl)acrylate (9c)
Formed as yellow crystals from n-hexane; yield (69%); M.P. 60˚C - 62˚C; IR (KBr) n∙cm−1 = 3447 (NH), 3098 (CH-arom), 2935 (CH-aliph), 1688 (C=O); 1H-NMR (DMSO-d6) d (ppm) = 1.09 (t, 3H, CH3, J = 12 Hz), 4.36 (q, 2H, CH2, J = 8 Hz), 6.78-8.04 (m, 9H, Ar-H + CH-oliffinic), 10.77 (s, 1H, NH); MS: m/z (%) 365 (M++2); Anal. Calcd. For C17H15Cl2N3O2 (363); C, 56.06; H, 4.15; N, 11.54; found C, 56.07; H, 4.16; N, 11.53.
2.9. General Procedure for the Preparation of Compound ( 13 a ,b)
A mixture of 8 (0.01 mol) and hydrazine hydrate, phenyl hydrazine (0.01 mol) in ethanol (30 mL) was heated under reflux for 12 hrs. The reaction mixture was allowed to cool and poured into crushed ice. The separated solid was filtered, washed with water and crystallized from the proper solvent to give 13a,b.
1) 4-(3-(4-Chlorophenyl)triaz-2-enyl)-1H-pyrazol-3-(2H)-one (13a)
Formed as yellow crystals from n-hexane; yield (69%); M.P. 122˚C - 124˚C; IR (KBr) n∙cm−1 = 3326, 3195, 3118 (3NH), 3066 (CH-arom), 1664 (C=O); 1H-NMR (DMSO-d6) d (ppm) = 5.19 (s, 1H, CH-pyrazole), 6.21 - 8.23 (m, 4H, Ar-H), 10.47 (s, 1H, NH), 10.53 (s, 1H, NH), 10.77 (s, 1H, NH); MS: m/z (%) 239 (M++2); Anal. Calcd. For C9H8ClN5O (237); C, 45.49; H, 3.39; N, 29.47; found C, 45.50; H, 3.38; N, 29.48.
2) 4-(3-(4-Chlorophenyl)triaz-2-enyl)-2-phenyl-1H-pyrazol-3-(2H)-one (13b)
Formed as yellow crystals from n-hexane; yield (69%); M.P. 100˚C - 102˚C; IR (KBr) n∙cm−1 = 3275, 3186 (2NH), 3060 (CH-arom), 1669 (C=O); 1H-NMR (DMSO-d6) d (ppm) = 5.74 (s, 1H, CH-pyrazole), 7.06 - 7.89 (m, 9H, Ar-H), 10.30 (s, 1H, NH), 11.40 (s, 1H, NH); MS: m/z (%) 313 (M+); Anal. Calcd. For C15H12ClN5O (313); C, 57.42; H, 3.86; N, 22.32; found C, 57.43; H, 3.87; N, 22.31.
2.10. 4-(3-(4-Chlorophenyl)triaz-2-enyl)isoxazol-5-(2H)-one (17)
A mixture of 8 (0.01 mol), hydroxylamine hydrochloride in glacial acetic acid (30 mL) containing anhydrous sodium acetate ( 1 g ) was heated under reflux for 24 hrs. The reaction mixture was allowed to cool and poured into cold water (60 ml). The separated solid was filtered and crystallized from ethanol to give brown crystals; yield (68%); M.p. 120˚C - 122˚C; IR (KBr) n∙cm−1 = 3446, 3149 (2NH), 2980 (CH-aliph), 1716 (C=O); 1H- NMR (DMSO-d6) d (ppm) = 6.90 - 7.92 (m, 7H, Ar-H, CH-isoxazole and 2NH); MS: m/z (%) 240 (M++2), Anal. Calcd. For C9H7ClN4O2 (238): C, 45.30; H, 2.96; N, 23.48; Found: C, 45.31; H, 2.97; N, 23.47%.
2.11. General Procedure for the Preparation of Compound ( 21 a ,b)
To boiling solution of compound 8 (0.01 mol), thiourea (0.01 mol) and guanidine hydrochloride in sodium ethoxide (30 ml). The reaction mixture was refluxed for 12 h, then allowed to cool and poured into crushed ice then acidified with HCl. The separated solid was filtered, washed with water and crystallized from the proper solvent to give 21 a ,b.
1) 5-(3-(4-Chlorophenyl)triaz-2-enyl)-2-thioxo-2,3-dihydropyrimidin-4-(1H)-one (21a)
Formed as yellow crystals from n-hexane; yield (69%); M.P. 102˚C - 104˚C; IR (KBr) n∙cm−1 = 3436, 3195 (2NH), 3094 (CH-arom), 1721 (C=O); 1H-NMR (DMSO-d6) d (ppm) = 7.23-7.84 (m, 5H, Ar-H + CH-pyrimi- dine), 8.60 (s, 1H, NH), 10.20 (s, 1H, NH), 12.20 (s, 1H, NH); MS: m/z (%) 283 (M++2); Anal. Calcd. For C10H8ClN5OS (281); C, 42.63; H, 2.86; N, 24.86; found C, 42.64; H, 2.87; N, 24.85.
2) 5-(3-(4-Chlorophenyl)triaz-2-enyl)-2-imino-2,3-dihydropyrimidin-4-(1H)-one (21b)
Formed as yellow crystals from n-hexane; yield (69%); M.P. 206˚C - 208˚C; IR (KBr) n∙cm−1 = 3445, 3421, 3372 (3NH), 3106 (CH-arom), 2942 (CH-aliph), 1690 (C=O); 1H-NMR (DMSO-d6) d (ppm) = 7.29 - 7.95 (m, 5H, Ar-H + CH-pyrimidine), 8.63 (s, 1H, NH), 10.00 (s, 1H, NH), 10.56 (s, 1H, NH), 10.82 (s, 1H, NH); MS: m/z (%) 266 (M++2); Anal. Calcd. For C10H9ClN6O (264); C, 45.38; H, 3.43; N, 31.75; found C, 45.39; H, 3.44; N, 31.74.
2.12. 5-(3-(4-Chlorophenyl)triaz-2-enyl)-6-oxo-2-thioxo-1,2,3,6-tetrahydropyridine- 3-carbonitrile (25)
A mixture of 8 (0.01 mol) and cyanothioacetamide (0.01 mol) in pyridine was heated under reflux for 12 hrs. The reaction mixture was allowed to cool and poured into crushed ice then acidified with HCl. The separated solid was filtered, washed with water and crystallized from ethanol to give brown crystals; yield (86%); M.p. 130˚C - 132˚C; IR (KBr) n∙cm−1 = 3447, 3419 (2NH), 3111 (CH-arom), 2202 (CN), 1718 (CO); 1H-NMR (DMSO-d6) d = 6.90 - 6.92 (d, 1H, CH-pyridine), 7.10 - 7.12 (d, 1H, =CH-pyridine), 7.23 - 8.52 (m, 4H, Ar-H), 10.30 (s, 1H, NH), 12.41 (s, 1H, NH); MS: m/z (%) 307 (M++2), Anal. Calcd. For C12H8ClN5OS (305): C, 47.14; H, 2.64; N, 22.91; Found: C, 47.15; H, 2.65; N, 22.90%.
3. Results and Discussion
3.1. Chemistry
The reaction of diazonium salts with proteins has been extensively used as a structural probe, typically in the elucidation of the topography of the active sites of enzymes [13] . The azo-proteins formed in this way arise largely from diazo-coupling with the activated aromatic rings of tyrosine and histidine residues and the ε-amino group of lysines [14] [15] . Thus, the reaction of aryldiazonium salts 2a with glycine ethyl ester hydrochloride in aqueous solution containing sodium acetate, the diazonium ion attacks at the NH2 moiety to affords stable triazene derivative 3a and shows no tendency to attack at the activated CH2, which would give rise to hydrozone formation [16] -[19] , based on its spectral data. For example, the 1H-NMR spectrum of compound 3a recorded in DMSO-d6 revealed the presence of a triplet signal at d = 1.17 ppm corresponding to methyl group, a quartet signal at d = 4.13 ppm corresponding to methylene group, a singlet signal at d = 5.02 ppm corresponding to methylene group, a multiplet signal at d = 6.53 - 8.22 ppm corresponding to aromatic protons and a singlet signal at d = 12.62 ppm corresponding to NH. The mass spectrum of the same product is in accordance with the proposed structure. It shows a very intense molecular ion peaks at 241 and a number of fragments agree with the proposed structure. Similarly, glycine ethyl ester coupled readily with aryl diazonium chlorides 2b-d in the same reaction conditions to give the corresponding triaza derivatives 3b-d. Cyclization of triaza derivatives 3a-c into triazol-5-ol derivatives 5a-c took place by fusion of 3a-c over melting point without solvent in the presence of ammonioum acetate for 20 min. Establishing of the compounds 5a-c were based on its elemental and spectral data. For example, the IR spectrum of compound 5a revealed the disappearance of carbonyl group. The 1H- NMR spectrum revealed the presence of a signal at d = 6.93 - 8.06 ppm corresponding to aromatic protons, CH-triazole and a singlet signal at d = 12.06 ppm corresponding to OH group. The mass spectrum of the same product is in accordance with the proposed structure as demonstrated in (Scheme 1).
The foregoing results prompt us to investigate the synthetic potentiality of stable triazenes 3a-d toward palmitoyl chloride. Thus, treatment of triazenes derivative 3a with palmitoyl chloride 6 in refluxing methylene chloride containing a catalytic amount of pyridine afforded ethyl-2-(3-(4-chlorophenyl)triaz-2-enyl)-3-oxooc- tadecanoate 7a [20] -[22] . The structure of 7a was established as a sole reaction product based on its spectral data. For example, the 1H-NMR spectrum of compound 7a recorded in DMSO-d6 revealed the presence of a triplet signal at d = 0.84 ppm corresponding to terminal three protons of long chain fatty acid, a multiblet signal at d = 1.22 - 1.28 ppm corresponding to (CH2) 12, a triplet signal at d = 1.56 ppm indicating methyl group, a triplet signal at d = 2.27 ppm corresponding to b-methylene group, a triplet signal at d = 2.48 ppm corresponding to a-methylene group, a singlet signal at d = 2.49 ppm corresponding to methine group, a quartet signal at d = 3.29 ppm corresponding to methylene group, a multiplet signal at d = 7.29 - 7.61 ppm corresponding to aromatic protons and a singlet signal at d = 9.94 ppm corresponding to NH. The mass spectrum of compound 7a revealed the molecular ion peak at m/z = 479 (M+) corresponding to C26HCl42N3O3. Similarly, the triazenes derivatives 3b-d was reacted with palmitoyl chloride in the same reaction conditions to afford 7b-d as demonstrated in Scheme 2.
Ethyl-2-(3-(4-chlorophenyl)triaz-2-enyl)acetate 3a was exploited as a key intermediate for the synthesis of hitherto unknown compounds through its reaction with some reagents. Thus, refluxing of compound 3a with dimethylformamide-dimethylacetal in dry dioxane for about 6 hrs afforded ethyl-2-(3-(4-chlorophenyl)triaz-2- enyl)-3-(dimethylamino)acrylate 8. Establishing structure 8 was based on its elemental analysis and spectral data. Thus, 1H-NMR spectrum of compound 8 revealed an intense singlet signal at d = 3.35 corresponding to two methyl functions, the oliffinic proton and NH group appeared as two singlet signals at d = 7.22 and 12.65 ppm

Scheme 1. Synthesis of compounds 1-5.
respectively. The mass spectrum of the same compound is in accordance with the proposed structure. Thus, it showed a very intense molecular ion peak at m/z = 296 and a number of fragments agree with the proposed structure. Further support of the structure 3 by its condensation with aromatic aldehydes. Thus, it was condensed with aromatic aldehydes in refluxing ethanol and in the presence of a catalytic amount of piperidine to give 9a-c. Establishing the structure of compound 9a-c. were based on its elemental analysis and spectral data. Thus, 1H-NMR spectrum of compound 9a as example revealed the absence of methylene function and revealed the presence of a singlet signal at d = 7.09 ppm corresponding to oliffinic proton (Scheme 3).
The enaminone 8 was examined as a key precursor toward a varity of nucleophilic reagents aiming at exploring its synthetic potentiality. Thus, when compound 8 is allowed to reflux with hydrazine and phenyl hydrazine in ethanol, pyrazolones 13a,b derivatives were obtained via the intermediacy of 10-12. Establishing structure 13a,b was based on its elemental analysis and spectral data. For example, The 1H-NMR spectrum of 13a revealed the presence of a singlet signal at d = 5.19 ppm corresponding to CH-pyrazole, a multiplet signal at d = 6.21 - 8.23 ppm corresponding to aromatic protons and the NH protons appeared as a singlet signal at d = 10.47, 10.53, 10.77 ppm. The mass spectrum of the same product is in accordance with the proposed structure. Compound 13a,b are assumed to be formed via the addition of the NH2 group of hydrazine, phenyl hydrazine to the double bond in compound 8 with subsequently elimination of dimethylamine molecule to give the non-isolable intermediate 11. Which underwent intramolecular cyclization to form the final product 13a,b through elimination of ethanol. When compound 8 was treated with hydroxylamine hydrochloride in refluxing glacial acetic acid, it afforded a single product identified as 4-(3-(4-chlorophenyl)triaz-2-enyl)isoxazol-5-(2H)-one 17. Formation of 17 from the reaction of 8 and hydroxyl amine is believed to be formed via initial addition of NH2 group to the double bond system of 8 to give the intermediate 14-16 by loss dimethylamine and ethanol to give isoxazole 17 as demonstrated in (Scheme 4).
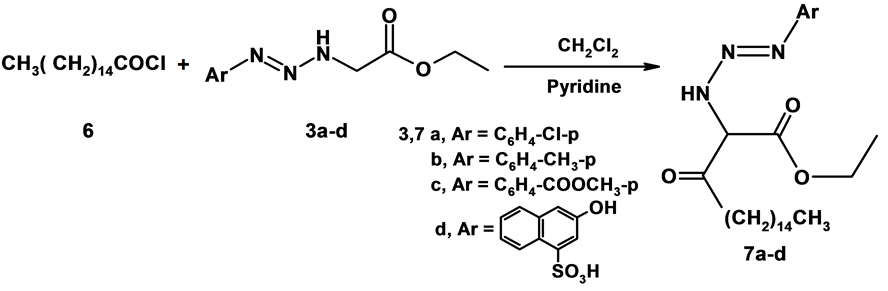
Scheme 2. Synthesis of compounds 6,7.
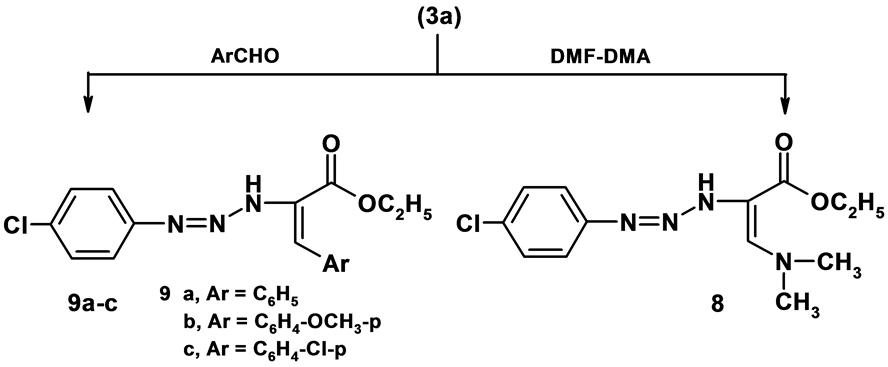
Scheme 3. Synthesis of compounds 8,9.
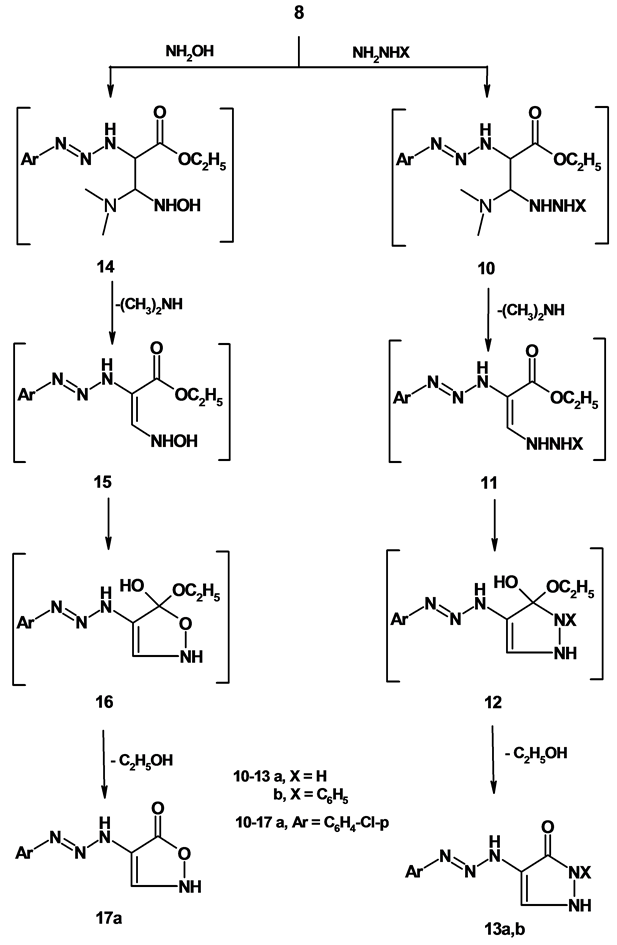
Scheme 4. Synthesis of compounds 10-17.
Treatment of compound 8 with thiourea in refluxing ethanol in the presence of sodium ethoxide, afforded 2-thioxo-2,3-dihydropyrimidin-4(1H)-one derivative 21a. Compound 21a is assumed to be formed via the addition of the NH2 group of thiourea to the activated double bond in compound 8 with subsequent elimination of dimethylamine molecule to give the non-isolable intermediate 19, which underwent intramolecular cyclization to form the final product 21a via loss of ethanol molecule. Also, the reaction of 8 with guanidine hydrochloride resulted in the formation imino pyrimidine derivative via the intermediacy of 18-20 according to their spectral analysis. For example, the IR spectrum of compound 21b revealed the presence of amino function at 3445, 3421, 3372 cm−1 and carbonyl function at 1690 cm−1. The 1H-NMR and mass spectrum of the same product are in accordance with proposed structure. Finally, compound 8 reacted also with cyanothioacetamide and gave the corresponding pyridinethione derivative 25. The structure of compound 25 was deduced from their elemental analysis and spectral data. The IR spectroscopic investigation of 25 revealed characteristic bands at 3447, 3419 cm−1 (2NH) and a pair of absorption band at 2202, 1718 cm−1 for (CN) and carbonyl group respectively, in addition, 1H-NMR of 25 revealed the presence of a doublet signal at d = 6.90 - 6.92 ppm corresponding to CH-pyridine, a doublet signal at d = 7.10 - 7.12 ppm corresponding to =CH-pyridine, a pair of signals at d = 10.30, 12.41 ppm corresponding to a pair of NH groups and a multiplet signal at d = 7.23 - 8.52 ppm corresponding to aromatic protons. The mass spectrum of compound 25 is in accordance with the proposed structure. Thus, it showed a very intense molecular ion peak at 305 (M+) and a number of fragment agree with the proposed structure, Formation of 25 from the reaction of 8 and cyanothioacetamide is believed to be formed via initial addition of the active methylene group of cyanothioacetamide on the double bond system of 8 with subsequent elimination of dimethylamine to give non-isolable intermediate 23 which underwent intramolecular cyclization to form the final product 25 via loss of ethanol molecules as demonstrated in Scheme 5.
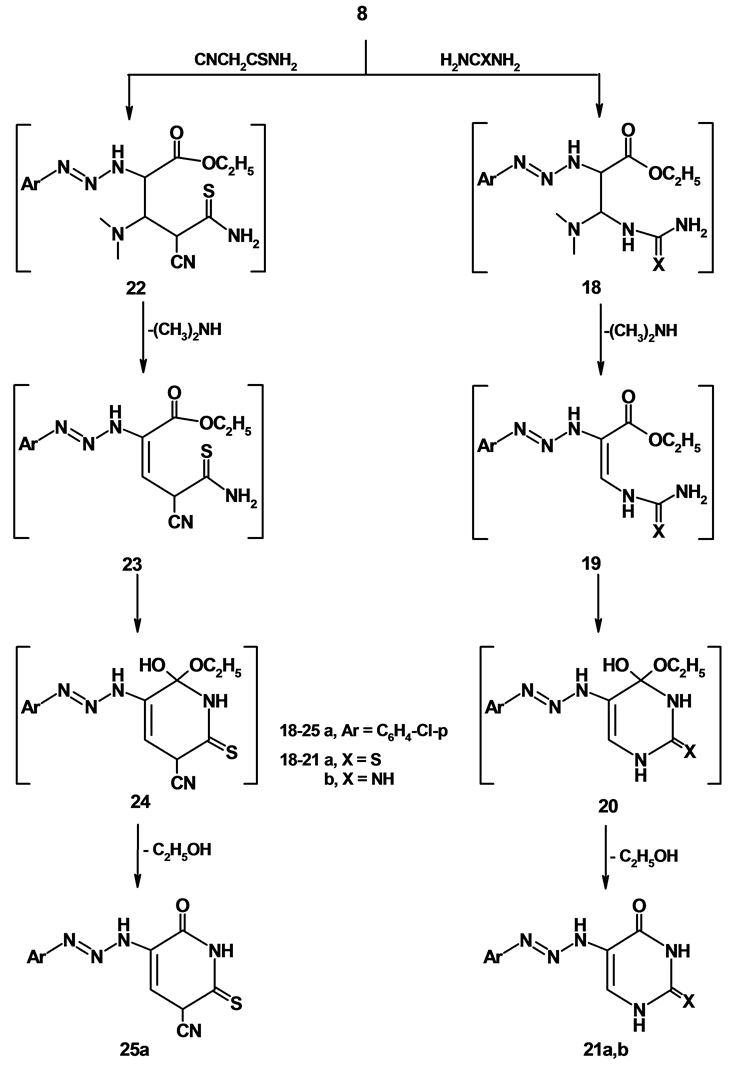
Scheme 5. Synthesis of compounds 18-25.
4. Conclusion
In conclusion, compounds 3a-d and 8 were used as efficient precursors for the synthesis of new heterocycles including triaz-2-enyl moiey with expected biological activities.
Cite this paper
Mohamed Ahmed Mahmoud AbdelReheim,Ahmed Mahmoud El-SayedTolba, (2016) Synthesis and Spectral Identification of Novel Stable Triazene: As Raw Material for the Synthesis Biocompatible Surfactants-Pyrazole-Isoxazole-Dihydropyrimidine-Tetrahydropyridine Derivatives. International Journal of Organic Chemistry,06,44-54. doi: 10.4236/ijoc.2016.61005
References
- 1. Piirma, I. (1992) Polymeric Surfactants, Surfactant Science Series. Marcel Dekker, New York.
- 2. Attwood, D. and Florence, A.T. (1983) Surfactant Systems. Their Chemistry, Pharmacy and Biology. Chapman and hall, London.
- 3. Fendler, J.H. (1982) Membrane Mimetic Chemistry. Characterizations and Applications of Micelles, Microemulsions, Monolayers, Bilayers, Vesicles, Host-Guest Systems, and Polyions. John Wiely & Sons, New York.
- 4. Infante, M.R., Pinazo, A. and Seguer, J. (1997) Non-Conventional Surfactants from Amino Acids and Glycolipids: Structure, Preparation and Properties. Colloide and Surfaces A: Physicochemical and Engineering Aspects, 49, 123-124.
- 5. Infante, M.R., Perez, L., Pinazo, A., Clapes, P., Moran, M.C., Angelet, M., Garcia, M.T. and Vinardell, M.P. (2004) Comptes Rendus Chimie, 7, 583-592.
http://dx.doi.org/10.1016/j.crci.2004.02.009 - 6. Moran, M.C., Pianzo, A., Perez, L., Clapes, P., Angelet, M., Garcia, M.T., Vinardell, M.P. and Infante, M.R. (2004) “Green” Amino Acid-Based Surfactants. Green Chemistry, 6, 233.
- 7. Xia, J., Qian, J. and Nnanna, I.A. (1996) Synthesis and Surface Properties of Amino Acid Surfactants from Industrial Waste Proteins. Journal of Agricultural and Food Chemistry, 44, 975-979.
http://dx.doi.org/10.1021/jf9505180 - 8. Xia, J., Xia, Y. and Nnanna, I.A. (1995) Structure-Function Relationship of Acyl Amino Acid Surfactants: Surface Activity and Antimicrobial Properties. Journal of Agricultural and Food Chemistry, 43, 867-871.
http://dx.doi.org/10.1021/jf00052a004 - 9. O’Dwyer, P.J., King, S.A., Plowman, J., Grieshaber, C.K., et al. (1988) Pyrazole: Preclinical Reassessment. Investigational New Drugs, 6, 305-310.
http://dx.doi.org/10.1007/BF00173649 - 10. Bekhit, A. and Abdel-Aziem, T. (2004) Design, Synthesis and Biological Evaluation of Some Pyrazole Derivatives as Anti-Inflammatory-Antimicrobial Agents. Bioorganic & Medicinal Chemistry, 12, 1935-1945.
http://dx.doi.org/10.1016/j.bmc.2004.01.037 - 11. Brogden, R.N. (1986) Pyrazolone Derivatives. Drugs, 32, 60-70.
http://dx.doi.org/10.2165/00003495-198600324-00006 - 12. Shishoo, C.J., Shirsath, V.S., Rathod, I.S. and Yande, V.D. (2000) Design, Synthesis and Antihistaminic (H1) Activity of Some Condensed 3-Aminopyrimidin-4(3H)-Ones. European Journal of Medicinal Chemistry, 35, 351-358.
http://dx.doi.org/10.1016/S0223-5234(00)00128-8 - 13. Stevens, M.F.G. (1976) The Medicinal Chemistry of 1,2,3-Triazines. Progress in Medicinal Chemistry, 13, 205-269.
http://dx.doi.org/10.1016/S0079-6468(08)70139-5 - 14. Tabachnik, M. and Sobotka, H. (1959) Azoproteins: I. Spectrophotometric Studies of Amino Acid Azo Derivatives. The Journal of Biological Chemistry, 234, 1726-1730.
- 15. Phillips Jr., J.H., Robrish, S.A. and Bates, C. (1965) High Efficiency Coupling of Diazonium Ions to Proteins and Amino Acids. The Journal of Biological Chemistry, 240, 699-704.
- 16. Daniels, T.A., Sidi, S. and Vaughan, K. (1977) Open-Chain Nitrogen Compounds. Part III. The Formation of Triazenes in the Reaction of Diazonium Salts with α-Aminoacetonitrile and Related α-Substituted Alky Lamines. Canadian Journal of Chemistry, 55, 3751-3754.
http://dx.doi.org/10.1139/v77-528 - 17. Baumgarten. R.J. (1967) Preparation of Ethyl Diazoacetate via a Triazene Intermediate. The Journal of Organic Chemistry, 32, 484-485.
http://dx.doi.org/10.1021/jo01288a059 - 18. Zahn, H., Wollrman, B. and Waschka, O. (1953) Reaktionen von Diazoniumsalzen mit Prolin, Cystein, Glycin und anderen Aminosauren. Zeitschrift fur physiologische Chemie, 294, 100.
http://dx.doi.org/10.1515/bchm2.1953.294.1.100 - 19. Shealy, Y.F. and Krauth, C.A. (1966) Imidazoles. II. 5(or 4)-(Monosubstituted Triazeno)imidazole-4(or 5)-carboxamides. Journal of Medicinal Chemistry, 9, 34.
http://dx.doi.org/10.1021/jm00319a008 - 20. Prakash, G.K. and Mahadevan, K.M. (2008) Enhancing the Properties of Wood through Chemical Modification with Palmitoyl Chloride. Applied Surface Science, 254, 1751-1756.
http://dx.doi.org/10.1016/j.apsusc.2007.07.137 - 21. Gerova, M., Rodrigues, F., Lamère, J.-F. and Dobrev, A. (2008) Self-Assembly Properties of Some Chiral N-Palmitoyl Amino Acid Surfactants in Aqueous Solution. Journal of Colloid and Interface Science, 319, 526-533.
http://dx.doi.org/10.1016/j.jcis.2007.12.004 - 22. Saqib, A., Afzal Pasha, M., Karigar, C. and Harish, M.S.N.R. (2010) Synthesis of Palmitic Acid Derivatives of P-Aminophenol and P-Aminobenzoic Acid with Improved Pharmacodynamic Profiles. Acta Pharmaceutica Sciencia, 52, 205-212.
NOTES

*Corresponding author.