Case Reports in Clinical Medicine
Vol.2 No.4(2013), Article ID:34934,5 pages DOI:10.4236/crcm.2013.24074
Clear cell sarcoma of the jejunum—Surgical management in two patients with review of the literature
![]()
1Department of Surgery, Loyola University Medical Center, Maywood, USA; anupama.mehta13@gmail.com
2Department of Surgery, Yale New Haven Hospital, New Haven, USA
3Department of Pathology, Loyola University Medical Center, Maywood, USA
4Department of Surgery, Cadence Healthgroup, Warrenville, USA
Copyright © 2013 Anupama Mehta et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received 24 April 2013; revised 26 May 2013; accepted 15 June 2013
Keywords: Clear Cell Sarcoma; Lymph Node Metastasis; Gastrointestinal Tract
ABSTRACT
Clear cell sarcoma is usually described as a malignant melanoma of the soft tissues. The overall prognosis is poor because of delay in diagnosis and vague clinical symptoms. It is rarely involved in the gastrointestinal tract, and its diagnosis is often missed secondary to infrequent occurrence and histological resemblance to melanoma. We present two cases of primary CCS of the jejunum whose clinical presentations were complicated by lymph node involvement. Prompt diagnosis and potential aggressive surgical intervention may improve overall survival.
1. INTRODUCTION
The original description of clear cell sarcoma (CCS) was reported by Enzinger in 1965 and has since been referred to as malignant melanoma of soft tissues [1]. While this rare and slow growing tumor typically occurs in the tendon sheaths and aponeuroses of distal extremities of adolescents and young adults, Ekfors et al. reported the first case of primary gastrointestinal CCS arising in the duodenum in 1993 [2].
This tumor’s involvement of the GI tract is rare and a source of diagnostic dilemmas considering that this peculiar sarcoma shows features of melanocytic differentiation [3]. Routine diagnostic testing relying on pure morphology and immunohistochemistry studies are insufficient for distinguishing between malignant melanoma and clear cell sarcoma of the GI tract [1]. Like CCS of the extremities, primary CCS of the GI tract presents as bundles or nests of pale staining eosinophilic spindle cells that are separated by dense fibrous tissue septa [3]. Similar to melanoma, CCS cells stain positive for S100 protein, contain melanosomes in more than half of cases, and often express antigens associated with melanin synthesis [4]. Covinksy et al. were the first to investigate molecular testing in order to distinguish between malignant melanoma and CCS [5]. CCS is genetically distinct from melanoma by nature of its lack of BRAF mutations [6]. Additionally, t(12;22)(q13;q12) is a unique translocation encoding the EWS/ATF1 fusion transcript found in more than three-fourths of CCS cases of the GI tract but absent in malignant melanoma [7].
The diagnosis of primary CCS of the GI tract is often overlooked because of this tumor’s infrequent involvement of the GI tract and its histologic resemblance to metastatic melanoma. As a result, optimal management, including extent of resection, is yet to be clearly defined. To date, there have been twenty-three cases of primary GI CCS that have been reported in the literature. In this case series, we illustrate two cases of primary CCS of the jejunum whose clinical presentation is complicated by the presence of lymph node involvement, further confounding the clinical behavior of this rare and perplexing clinical entity.
2. CASE REPORTS
2.1. Case 1
A twenty-nine years old female without past surgical history presented with a four year history of escalating nausea, vomiting, and abdominal pain was admitted with the diagnosis of small bowel obstruction. Abdominal computed tomography (CT) scan revealed bulky mesenteric adenopathy and a large, circumferential mass compressing the mid to distal small bowel. She underwent an exploratory laparotomy with small bowel resection, lymphadenectomy, primary anastomosis, and left ovarian dermoid cystectomy. Her final pathology was consistent with CCS.
During routine follow-up 3 months later, two low attenuating liver lesions measuring 1.2 cm were seen in Segments IV and V on CT but did not demonstrate increased activity utilizing Positron Emission Tomography (PET). She subsequently underwent radiofrequency ablation at an outside facility. Seven months after initial diagnosis, new liver lesions were noted in Segments II, III, and IV on CT. A CT scan performed eleven months after her initial diagnosis revealed six hypo-attenuating liver lesions and four of the six lesions demonstrated PET activity. At exploration, each lobe demonstrated three lesions. A formal right hepatectomy was performed with microwave ablation for the left sided lesions. Final pathology revealed metastatic CCS. Postoperatively, she was started on a chemotherapy regimen consisting of adriamycin and ifosfamide after developing ascites and recurrence as revealed on CT imaging. While this regimen led to stabilization of liver lesions, interval imaging demonstrated intra-abdominal metastases with omental disease and worsening ascites. She died 23 months after her initial diagnosis.
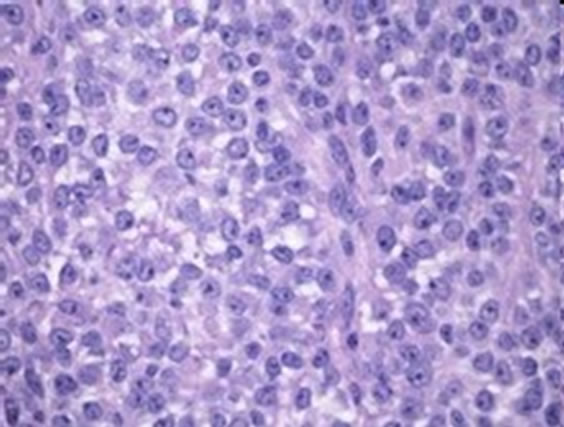
Histopathology Initial specimen A 25 cm segment of the small intestine was resected with 8 cm proximal and 15 cm distal margins negative for tumor. The jejunal mass was noted to be 4.0 × 3.0 × 2.5 cm in size and 5 of the 22 lymph nodes were positive. Histological examination revealed an epithelioid neoplasm arranged in nests separated by fibrovascular septa. The cells had cord like arrangement and vacuolated cytoplasm. There were enlarged nuclei with vascular chromatin and prominent nucleoli with easily seen mitotic figures (Figures 1(a) and (b)). Immunohistochemical markers were positive for synaptophysin, neuron specific enolase, S100, and Melan A. Markers tested for tyrosinase, MART-1, HMB45, chromogranin, AE1/3 cytokertain, SK8/18 cytokeratin, CD34, c-kit, smooth muscle actin and desmin were all negative. Fluorescence in situ hybridization (FISH) was performed to exclude the possibility of metastatic melanoma. The t(12;22)(q13;q12) translocation characteristic of CCS was present.
2.2. Case 2
A twenty-four years old female with a medical history significant for leukemia treated at age six, depression, and seizure disorder, initially reported by Venkataraman
 (a)
(a)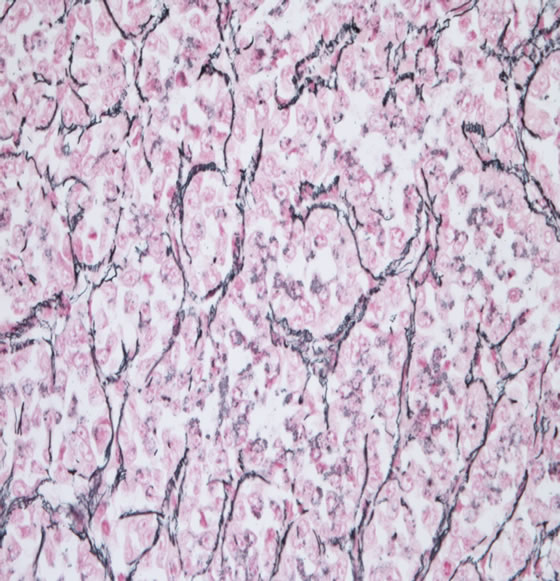 (b)
(b)
Figure 1. (a) Compact nests of rounded cells with clear cytoplasm. Nuclei are round with vesicular chromatin and prominent nucleoli; (b) Nesting pattern of growth defined by reticulin stain Cells are arranged in nest like pattern with fibrovascular cords.
el al., presented with unintentional 12 kg weight loss over six months and four days of nausea, vomiting, and abdominal pain [8]. CT scan demonstrated a 1.9 cm thickening of the distal jejunum and proximal ileum with adjacent mesenteric lymphadenopathy. Diagnostic testing with esophagogastroduodenoscopy (EGD) and colonoscopy proved normal, but small bowel follow through revealed a polypoid lesion in the distal small bowel and subsequent capsule endoscopy resulted in a trapped capsule at the site of obstruction. She underwent exploratory laparotomy, small bowel resection with primary anastomosis and lymphadenectomy. Her pathology was consistent with CCS.
After a disease free interval of three years, surveillance CT revealed a 2 cm right sided liver lesion. She underwent a partial right hepatic lobectomy that revealed metastatic CCS with negative margins. Additional right sided liver lesions were demonstrated five months later on CT scan and were confirmed on PET scan. Then she underwent a completion formal right hepatectomy. Surveillance CT three months after her second hepatic resection indicated a growing nodule in her left lung measuring 0.4 cm and an interval increase in aortocaval lymph nodes, all of which were PET avid. Because these findings were concerning for further metastatic disease, she underwent and completed a course of adriamycin and ifosfamide which resulted in initial regression.
However, within months of initial discovery both the lung nodule and the aortocaval lymphadenopathy demonstrated enlargement. She underwent bronchoscopy with robotic assisted resection of the left lower lobe of the lung and aortocaval lymph node dissection revealing a 2.3 × 2.0 × 1.5 cm mass consistent with CCS, focal infiltration of the bronchial wall, and 3 positive lymph nodes. Three months following these surgical interventions, she developed a 1.2 × 2.6 cm retroperitoneal soft tissue mass medial to the IVC with corresponding increased metabolic activity on PET scan. She was placed on cisplatin and combretastatin therapy and terminated treatment secondary to its adverse side effects. Follow-up CT scan revealed a new periportal soft tissue lesion adjacent to the caudate lobe of the liver and an interval increase in size of the original retroperitoneal mass. She underwent stereotactic body radiotherapy with reduction in nodal size.
Seven and a half years after initial diagnosis, she had disease recurrence in the chest cavity. CT scan revealed a growing lesion near the hilum of the right middle lobe and a soft tissue mass adjacent to the suture line and left diaphragmatic margin. She had a right middle lobectomy with mediastinal lymph node dissection, left VATS with decortication of the left lung and extended resection of the left lower lobe, diaphragmatic repair, en block pericardial resection, and resection of the left posterior pleural mass. The right middle lobe, left pleura, and left lower lobe wedge with pericardial resection specimens were positive for CCS and margins were negative. This patient elected to undergo stereotactic radiosurgery in lieu of surgical re-excision when she presented with pulmonary and hepatic recurrent disease just 3 months later. Follow-up imaging indicated further hepatic metastases and new brain metastasis and underwent radiation therapy for her brain metastasis. She died 8 years after initial diagnosis.
Histopathology Initial specimen A 27 cm segment of small bowel was excised with 15 cm proximal and 12 cm distal margins both free of tumor. The jejunal mass was 5 × 2 × 1.5 cm in size and three of the twenty-two lymph nodes were positive. Pathology was consistent with large cell malignant neoplasm with epitheliod and neuroendocrine features. Immunohistochemical markers were positive for S100 and pankeratin. Markers were negative for synaptophysin, Melan A, tyrosinase, MART-1, HMB45, chromogranin, CD34, c-kit, smooth muscle actin, desmin, INHIBIN, CK20, CK7, LCA, Vimentin, and BCL2. FISH was positive for t(12;22)(q13;q12) translocation, consistent with clear cell sarcoma. KIT and CD34 were both negative which excluded diagnosis of an epithelioid gastrointestinal stromal tumor.
3. DISCUSSION
Although there have been reports of long term survivors diagnosed with CCS, the overall prognosis is poor for several reasons. First, the initial diagnosis of gastrointestinal CCS proves to be challenging given its rare occurrence. In the current literature, there are only twenty-four cases of gastrointestinal CCS reported, including the two presented here (Table 1). Of these, seventeen (70.8%) arose from the small bowel and nine (37.5%) involved the jejunum specifically. The duration of symptoms varies from patient to patient, and most patients experience some delay in diagnosis secondary to the vague and nonspecific nature of their symptoms. Second, the morphologic and immunohistochemical features of CCS are strikingly similar to that of melanoma making it difficult to distinguish between these two disparate tumors. Third, gastrointestinal CCS has an aggressive clinical course with a high incidence of regional and distant metastases. Lymph node involvement was reported in 64.7% of patients diagnosed with gastrointestinal CCS. Those with jejunal CCS had lymph node involvement 80% of the time. Metastatic disease to the liver was reported in 60% of patients with gastrointestinal CCS and in 50% of those with jejunal CCS.
We identified two cases at our institution of morphologically, immunohistologically, and genetically proven primary jejunal CCS. Each subsequently developed metastatic disease to both the liver and lymph nodes, confounding the typical behavior of sarcomas that infrequently metastasize to lymph nodes. These are the only two case reports of jejunal CCS with spread to both lymph nodes and the liver. The importance of prompt diagnosis is apparent from these case reports. Case 2 was diagnosed within six months of symptom development compared to the four years that Case 1 experienced before being diagnosed with CCS. The disease free interval following diagnosis for Case 2 was three years compared to eleven months for Case 1, and Case 2 lived approxi-


Table 1. Case reports of clear cell sarcoma of small bowel with molecular confirmation.
mately two years longer than Case 1 potentially because of expedient diagnosis and surgical intervention.
4. CONCLUSION
CCS remains a diagnostic and treatment dilemma. Given the infrequent occurrence, large studies identifying optimal patient approach are lacking. As is the case with other retroperitoneal sarcoma types, it would seem that aggressive surgical resection remains a mainstay in treatment. Despite aggressive surgical resections, the inherent natural biology of CCS seems to portend a poor prognosis with biologic activity that, in the current understanding, remains relatively unpredictable as evidenced by the disparate metastatic behavior. The role of systemic therapy, however, remains unanswered. Optimal treatment protocols for gastrointestinal CCS will remain a significant challenge given the rarity of this tumor type.
![]()
![]()
REFERENCES
- Lyle, P.L., Amato, C.M., Fitzpatrick, J.E., et al. (2008) Gastrointestinal melanoma or clear cell sarcoma? Molecular evaluation of 7 cases previously diagnosed as malignant melanoma. The American Journal of Surgical Pathology, 32, 858-866. doi:10.1097/PAS.0b013e31815b8288
- Ekfors, T.O., Kujari, H. and Isomaki, M. (1993) Clear cell sarcoma of tendons and aponeuroses (malignant melanoma of soft parts) in the duodenum: The first visceral case. Histopathology, 22, 255-259. doi:10.1111/j.1365-2559.1993.tb00115.x
- Taminelli, L., Zaman, K., Gengler, C., et al. (2005) Primary clear cell sarcoma of the ileum: An uncommon and misleading site. Virchows Archiv, 447, 772-777. doi:10.1007/s00428-005-0019-y
- Rosai, J. (2005) Editorial: Clear cell sarcoma and osteoclast-rich clear cell sarcoma-like tumor of the gastrointestinal tract: One tumor type or two? Melanoma or sarcoma? International Journal of Surgical Pathology, 13, 309-311. doi:10.1177/106689690501300401
- Covinsky, M., Gong, S., Rajaram, V., et al. (2005) EWSATF1 fusion transcripts in gastrointestinal tumors previously diagnosed as malignant melanoma. Human Pathology, 36, 74-81. doi:10.1016/j.humpath.2004.10.015
- Antonescu, C.R., Nafa, K., Segal, N.H., Dal Cin, P. and Ladanyi, M. (2006) EWS-CREB1: A recurrent variant fusion in clear cell sarcoma—association with gastrointestinal location and absence of melanocytic differentiation. Clinical Cancer Research, 12, 5356-5362. doi:10.1158/1078-0432.CCR-05-2811
- Lagmay, J.P., Ranalli, M., Arcila, M., et al. (2009) Clear cell sarcoma of the stomach. Pediatric Blood & Cancer, 53, 214-216. doi:10.1002/pbc.22014
- Venkataraman, G., Quinn, A.M., Williams, J. and Hammadeh, R. (2005) Clear cell sarcoma of the small bowel: A potential pitfall. APMIS, 113, 716-719. doi:10.1111/j.1600-0463.2005.apm_243.x
- Granville, L., Hicks, J., Popek, E., et al. (2006) Visceral clear cell sarcoma of soft tissue with confirmation by EWS-ATF1 fusion detection. Ultrastructural Pathology, 30, 111-118. doi:10.1080/01913120500406400
- Achten, R., Debiec-Rychter, M., De Wever, I., et al. (2005) An unusual case of clear cell sarcoma arising in the jejunum highlights the diagnostic value of molecular genetic techniques in establishing a correct diagnosis. Histopathology, 46, 464-473. doi:10.1111/j.1365-2559.2005.02010.x
- Adair, C., Ro, J.Y., Sahin, A.A., et al. (1994) Malignant melanoma metastatic to gastrointestinal tract: A clinicopathologic study. International Journal of Surgical Pathology, 2, 3-10. doi:10.1177/106689699400200102
- Comin, C.E., Novelli, L., Tornaboni, D., et al. (2007) Clear cell sarcoma of the ileum: Report of a case and review of literature. Virchows Archiv, 451, 839-845. doi:10.1007/s00428-007-0454-z
- Donner, L.R., Trompler, R.A. and Dobin, S. (1998) Clear cell sarcoma of the ileum: The crucial role of cytogenetics for the diagnosis. American Journal of Surgical Pathology, 22, 121-124. doi:10.1097/00000478-199801000-00016
- Fukuda, T., Kakihara, T., Baba, K., Yamaki, T., Yamaguchi, T. and Suzuki, T. (2000) Clear cell sarcoma arising in the transverse colon. Pathology International, 50, 412- 416. doi:10.1046/j.1440-1827.2000.01066.x
- Joo, M., Chang, S.H., Kim, H., et al. (2009) Primary gastrointestinal clear cell sarcoma: Report of 2 cases, one case associated with IgG4-related sclerosing disease, and review of literature. Annals of Diagnostic Pathology, 13, 30-35. doi:10.1016/j.anndiagpath.2008.10.003
- Kosemehmetoglu, K. and Folpe, A.L. (2010) Clear cell sarcoma of tendons and aponeuroses, and osteoclast-rich tumor of the gastrointestinal tract with features resembling clear cell sarcoma of soft parts: A review and update. Journal of Clinical Pathology, 63, 416-423. doi:10.1136/jcp.2008.057471
- Pacheco, M., et al. (2010) Small blue round cell tumor of the interosseous membrane bearing a t(2;22)(q34;q12)/ EWS-CREB-1 translocation: A case report. Molecular Cytogenetics, 3, 12. doi:10.1186/1755-8166-3-12
- Pauwels, P., Debiec-Rychter, M., Sciot, R., et al. (2002) Clear cell sarcoma of the stomach. Histopathology, 41, 526-530. doi:10.1046/j.1365-2559.2002.01509.x