Advances in Bioscience and Biotechnology
Vol.4 No.1(2013), Article ID:27233,6 pages DOI:10.4236/abb.2013.41011
Effect of pyk gene location on constructing dATP conversion E. coli strain
![]()
State Key Laboratory of Bioreactor Engineering, East China University of Science and Technology, Shanghai, China
Email: *jbao@ecust.edu.cn
Received 18 November 2012; revised 30 December 2012; accepted 10 January 2013
Keywords: pyk Gene Location; dATP Conversion Strain; 5-Untranslation Regions
ABSTRACT
A dATP conversion E. coli strain could be constructed when both pyk and adk1 gene were expressed successfully. pyk gene encodes pyruvate kinase (PK) could be expressed, when inserted it before adk1 gene which encodes adenylate kinase (AK) in plasmid pET-pyk-adk1 after transform into E. coli and the recombinant could be used to convert dATP from dAMP. Another plasmid pET-adk1-pyk, which inserted pyk gene behind of adk1, the recombinant E. coli transformed with this plasmid could not convert dAMP into dATP, pyk gene cannot be translated in this recombinant. The different translation levels of pyk with gene location switching caused mainly by the different secondary structures formed by the 5’-untranslation regions and the gene sequence of its 5’-terminal. The dATP product E. coli strain could be constructed when cloned pyk gene at an optimum location.
1. INTRODUCTION
Deoxyadenosine triphosphate (dATP) is one of the necessary precursors used in DNA biosynthesis, especially in the PCR reaction. The commercial dATP products come mainly from chemical synthesis, but the chemical method gives relatively low yield and need the toxic substances issued by the Environmental Protection Agency, like pyridine and dimethylformamide is cited from [1]. The biosynthesis method is environmental friendly one, a couple phosphorylation reactions can be used to synthesis dATP for dAMP. In the first step, adenylate kinase (AMP kinase, AK) was added to convert deoxynucleoside monophosphates (dAMP) into deoxynucleoside diphosphates (dADP), and then pyruvate kinase (PK) participated in subsequentlly to convert deoxynucleoside diphosphates (dADP) into deoxynucleoside triphosphates (dATP) [1]. People should express and purify two enzymes individually, this complicated the procedure and increased the cost.
To clone two or more genes into one host was feasible in recently. In order to simplify the biosynthesis process, two kinases were cloned onto a high copy plasmid pET- 17b and performed overexpression in E. coli BL21. The recombinants are used to catalyze dATP from dAMP after inducting protein expression of plasmid pET-pykadk1. However, the poor expression spectrum of pyk gene on the pET-adk1-pyk plasmid prevented the conversion of dAMP to dATP, and leading to the accumulation of intermediary dADP produced.
There are some reports on the relationship of gene location and the translation efficiency [2], the transcript level and mRNA stability of the target gene are thought important factors on gene expression efficiency, and the 5’ untranslated region is also an important factor on protein expression efficiency [3]. As for a specific gene, the story remains incomplete, the factors mentioned above would have different contributions [2,4].
As for the pyk gene, the possible reasons were clarified in this report for its different expression efficiency after changing the location with adk1 on plasmid pET- 17b. Except the different secondary structures of its mRNA formed with the 5’-UTR, the transcript level and mRNA half-time were remained same level in the two recombinants. The solo difference of pyk mRNA secondary structures seems a main reason on PK expression level in E. coli BL21 (pET-adk1-pyk). The different secondary structure afford pyk mRNA a higher Minimum Gibbs free energy values (ΔG, –26.7 kJ·mol–1) in E. coli BL21 (pET-adk1-pyk) than that in BL21 (pET-adk1-pyk) (ΔG, –18.0 kJ·mol–1).
2. MATERIAL AND METHODS
2.1. Reagents and Strains
Restriction endonucleases, T4 DNA ligase and Pfu DNA polymerase were purchased from Fermentas (HanoverUSA); Isopropyl-beta-D-thiogalactopyranoside (IPTG) was from Sigma-Aldrich (St Louis, MO, USA); Diaflo Ultrafilter membrane and RNA extracted Kit were from Omega Biotech (Vancouver, Canada); Ampicillin and LB broth were from Sangon Biotech (Shanghai, China).
The E. coli BL21 (DE3) strain 1 was purchased from Novagen, and the plasmid pET-jb was constructed in our lab as reported [1], per liter LB medium contains 100 mg·ml–1 of ampicillin.
2.2. Plasmid Construction and Protein Expression
The pyk gene was amplified from Bacillus sp. ATCC 31821 by PCR according to the gene sequence of NCBI database (accession No: gi 255767013). The primers sequences were: TAGTCGACAAGAAGGAGAACAAATGAGAAAAAC (forward with Sal I and ribosome binding site (RBS) region), and CACCTCGAGTAATTAAA GAACGCT (reverse with Xho I) synthesized by Shanghai Biotech (Shanghai, China). pET-pyk-adk1 and pET-adk1-pyk was constructed by replacing the amplified fragment at the Sal I and Xho I sites in pET-jb1 with adk1 [1]. The endonuclease sequences and the RBS sequence were underlined and framed.
After confirmed by DNA sequence, the plasmids were transformed into E. coli BL21 (DE3), and named as E. coli BL21 (pET-adk1-pyk) and E. coli BL21 (pETpyk-adk1). The co-expression of PK and AK was conducted under optimum inducing conditions, and used for dAMP conversion and protein spectrum analyses on 10% SDS-PAGE. E. coli BL21 (DE3) harboring plasmid pET-17b was set as control.
2.3. Conversion Analysis of Recombinants
The conversion activity of the recombinants was conducted in separate but analogous reactions. It containing 100 ml reaction buffer (50 mM Tris buffer pH 8.0 with 500 mM dAMP, 1000 mM PEP, 100 mM KCl, and 10 mM MgCl2), added 2 mg cell pellets of the recombinant (collected from 50 ml culture with OD600 at 2.0), the mixtures were kept at 30˚C for 2 hrs. The reactions course was monitored periodically and detected using high performance liquid chromatography (HPLC, LC-9A, Shimadzu). Using Intensil ODS-2 column (4.6 mm × 150 mm, GL Sciences, 254 nm), the elution buffer was 10 mM KH2PO4 and 10 mM K2HPO4 (pH 7.0) remained an elution rate of 1.0 ml/min at ambient temperature.
2.4. pyk mRNA Quantify and Stability Assay
Collected the engineered E. coli at OD600 reached 0.8, extract and purify the RNA using RNA extracted Kit from Omega Biotech, about 25 μg RNA was extracted in this test. The hybridization and detection procedure were following the method of Ravanshad [5]. The cDNA probe sequence of pyk was GCGAATACGCCAGATACGAG with the 5’-end Dig labeled, according to pyk gene sequence of NCBI database (gi 255767013). The optical density of the hybrid samples was determined, BL21 (DE3) harboring plasmid pET-17b was set as control.
Actinomycin D was used as transcription inhibitor for mRNA half-life detection. The optimal concentration of Actinomycin D was from 0.05 to 10 µg·ml–1 added in the culture, after 20 minutes, cells were collected and used for dot blot test. The degradation rate of pyk mRNA was tested at different times 0 min - 60 min within an optimal Actinomycin D concentration.
2.5. pyk mRNA Secondary Structure Prediction
The secondary structure of the pyk mRNA was predicted using online software MFOLD (http://mfold.bioinfo.rpi.edu/cgi-bin/rna-form/cgi) based on the principle of minimum free energy [6]. The mRNA sequence calculated was the +25 bp fragment of the 5’-UTR (Untranslated Region) including the RBS sequence and the first 11 codons started from AUG.
3. RESULTS AND DISCUSSION
3.1. Recombinant Plasmid Construction and PK Expression Analysis
There are two castles of adk1 and pyk gene with location changing under the same operon controlling, one is 5’- adk1-pyk-3’ and another is 5’-pyk-adk1-3’. Two plasmids pET-pyk-adk1 and pET-adk1-pyk were constructed accordingly, as Figures 1(a) and (b) show. After being transformed into BL21 (DE3), two recombinants BL21 (pET-pyk-adk1) and BL21 (pET-adk1-pyk) were named.
The protein expression was conducted under different IPTG induction concentration (0, 0.2, 0.4, 1.0 mM) (Figure 2(a)) and time course (0, 2, 4, 8 hrs) (Figure 2(b)). The SDS-PAGE for E. coli BL21 (pET-pyk-adk1) showed two protein bands at 26 kD and 58 kD positions [1], AK and PK were expressed successfully in this strain. However, the lysate of E. coli BL21 (pETadk1-pyk) has only a 26 kD band of AK, though under the same induction conditions as E. coli BL21 (pETpyk-adk1) (Figures 2(a) and (b)).
3.2. dATP Conversion Activity of the Two Recombinants
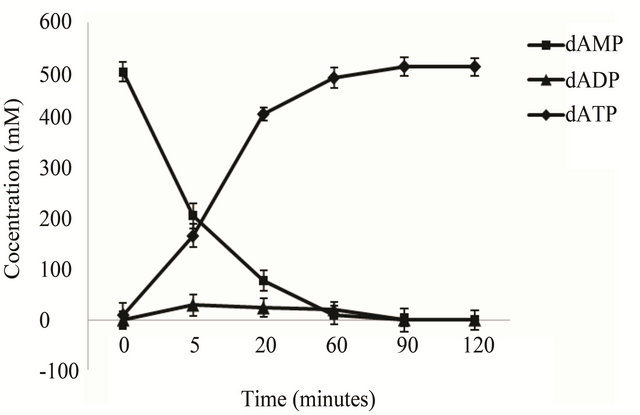
Under the given reaction conditions, E. coli BL21 (pETpyk-adk1) 500 mM dAMP substrates could be converted into dATP within 90 minutes and no by-product as

(a) (b)
Figure 1. Physical map of the co-expression plasmids pET-adk1-pyk (a) and pET-pyk-adk1(b) Denote: pBR322 ori, origin of replication; Ampr, ampicilinresistant marker.
 (a)
(a)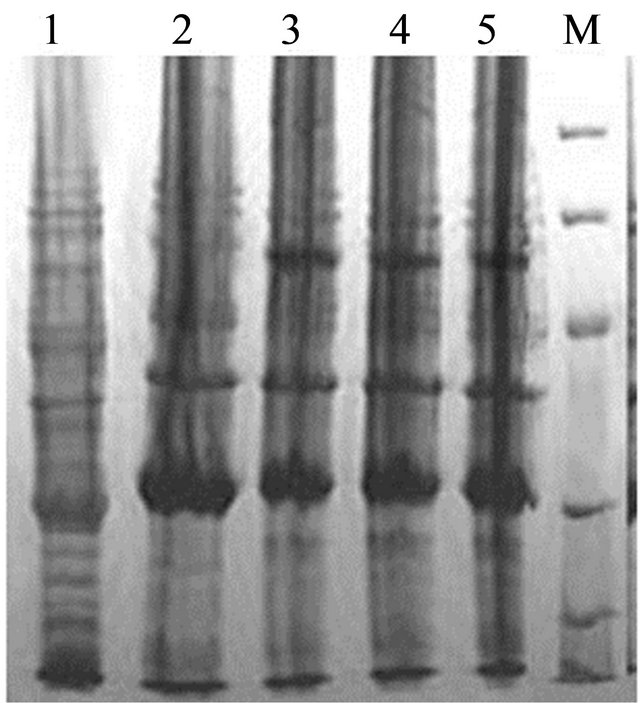 (b)
(b)
Figure 2. SDS-PAGE of AK and PK in the recombinant strains E. coli BL21 (pET-adk1-pyk) and E. coli BL21(pET-pyk-adk1) under different IPTG concentration at 37 ˚C for 6 hours (a) and induction time with 0.4 mM IPTG in the medium at 37˚C (b). (a) Lane 1: Negative control; Lane 2 to 5 and Lane 6 to 9: protein spectrum with 0, 0.2, 0.4, 1.0 mM IPTG in E. coli BL21 (pET-pyk-adk1) and E. coli BL21 (pET-adk1-pyk) respectively; Lane 10: Protein MW markers. (b) Lane 1: Negative control; Lane 2 to 5 and Land 6 to 9: protein spectrum after inducing for 0, 2, 4, 8 hours with 0.4 mM IPTG for E.coli BL21 (pET-pyk-adk1) and E. coli BL21 (pET-adk1-pyk) respectively; Lane 10: protein MW markers.
(Figure 3(a)) shows. When use the recombinant E. coli BL21 (pET-adk1-pyk) as catalyst, a bound up of dADP accumulated in the reactor (Figure 3(b)) , the conversion reaction did not go to completion.
3.3. pyk mRNA Transcript Level and Stability Analysis in the Two Strains
The optical density of pyk mRNA hybrid blot with the cDNA probe in E. coli BL21 (pET-adk1-pyk) (dot 2) and E. coli BL21 (pET-pyk-adk1) (dot 3) are comparable, both stronger than in E. coli BL21 (pET-17b) (dot 1) as Figure 4 shows.
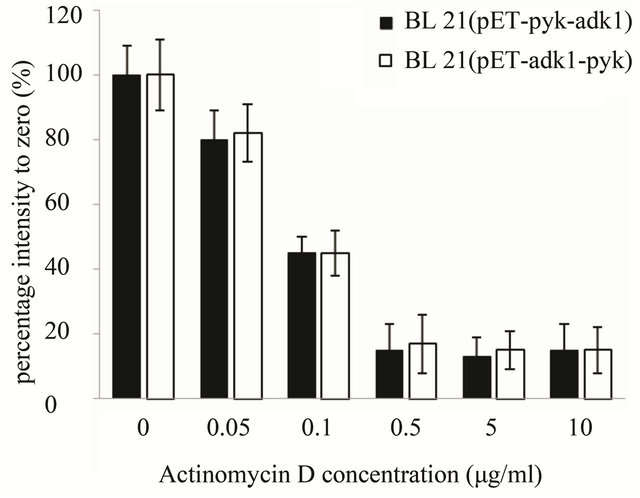
As for the stability analysis of pyk mRNA, with Actinomycin D concentration increasing the optical intensity of the hybrid blots decreasing in both recombinants accordingly. The intensity of the hybrid dot was only about 15% with 0.5 µg·ml–1 Actinomycin D in the medium, the transcript activity of pyk was almost inhibit completely in both strains (Figure 5(a)). 10 minutes later with 0.5 μg·ml–1 Actinomycin D in the medium, the intensity of pyk hybrid blot in the two recombinants was decreased to 16% of the zero point (Figure 5(b)) and kept at the same level, it was very weak in the two strains. The results indicated that the pyk gene has the same transcript level and mRNA stability in the two recombinants.
3.4. pyk mRNA Secondary Structure Prediction
The sequence of of pyk mRNA which used for secondary structures prediction including the 25 nucleotides of
 (a)
(a)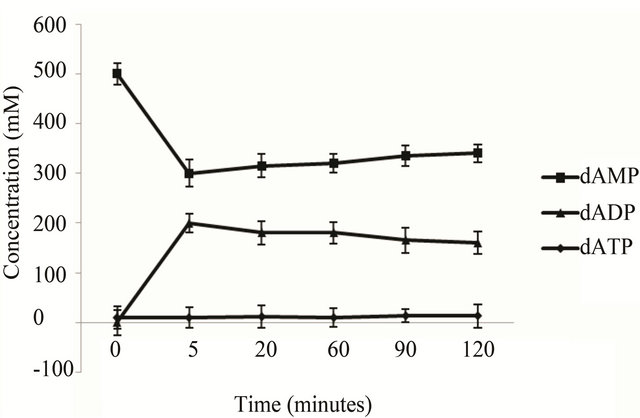 (b)
(b)
Figure 3. dATP conversion time course of the engineering strains. The catalyst is 2 ml cell lysate (collected from 50 ml culture at OD600 2.0) of the E. coli BL 21 (pET-pyk-adk1) (a) E. coli BL 21 (pET-adk1-pyk) (b), the reaction mixed with 500 mM of dAMP, 1000 mM PEP at 30˚C in 50 mM Tris at pH of 8.0, with 100 mM potassium chloride and 10 mM magnesium chloride.
1 2 3
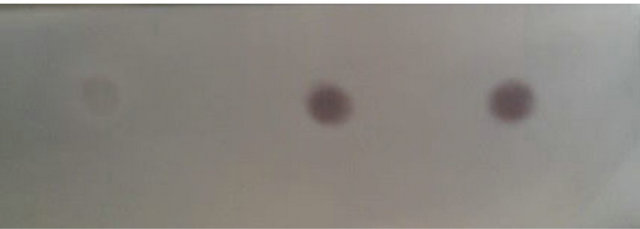
Figure 4. pyk gene transcript level assay in E. coli BL21 (pET-adk1-pyk) and E. coli BL21 (pET-pyk-adk1) using Dot-blot tests. Hybridized blot with pyk cDNA probe in E. coli pET-17b Dot 1, in E. coli BL21 (pET-adk1-pyk) Dot 2 and in E. coli BL21 (pET-pyk-adk1) Dot 3.
 (a)
(a)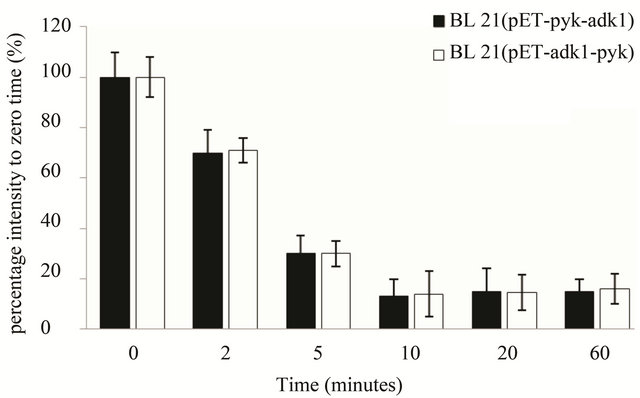 (b)
(b)
Figure 5. pyk mRNA stability assay in E. coli BL21 (pET-adk1-pyk) and E.coli BL21 (pET-pyk-adk1). (a) add 0; 0.05; 0.1; 0.5; 5.0; 10.0 µg ml-1 Actinomycin D for 20 minutes before collected for Dot blot test; 0 µg ml−1 Actinomycin D inhibition rate was set as 100%. (b) pyk mRNA degradation rate at different time point (0; 2; 5; 10; 20; 40; and 60 min) with 0.5 µg·ml-1 Actinomycin D in the medium. 0 point was set as 100%.
5’-UTR and the first 11 codon from ATG start codon, the secondary structures in E. coli BL21 (pET-adk1-pyk) and E. coli BL21 (pET-pyk-adk1) are shown in Figure 6. The pyk mRNA 5’-end formed more hairpin structures (Figure 6(a)) in E. coli BL21 (pET-adk1-pyk) than it did in E. coli BL21 (pET-pyk-adk1) (Figure 6(b)). And the calculated Gibbs free energy values (ΔG) of pyk mRNA secondary structure based on the principle of minimum free energy in E. coli BL21 (pET-pyk-adk1) was –26.7 kJ·mol–1, it was –18.0 kJ·mol–1 in E. coli BL21 (pETadk1-pyk). There were 31.8% energy barriers increased in BL21 (pET-pyk-adk1) than in BL21 (pET-adk1-pyk) as Table 1 show.
4. DISCUSSION
To simplify the biosynthesis procedure of dATP from dAMP, an efficient method was reported here. Two phosphorylate kinases were coexpressed in the same engineered bacteria, adenylate kinase (AK) which converted deoxynucleoside monophosphates (dAMP) to deoxynucleoside diphosphates (dADP) encoded by adk1- gene, and pyruvate kinase (PK) participating in the sequential step to convert the intermediate product deoxynucleoside diphosphates (dADP) to deoxynucleoside triphosphates (dATP) encoded by the pyk gene.
Though there have two kinds of arranging castle for pyk gene and adk1 gene, only the recombinant E. coli BL21 (pET-pyk-adk1) could be used to catalyze 500 mM of dAMP converting to dATP. Another recombinant E. coli BL 21(pET-adk1-pyk) could not complete this coupling phosphorylation reaction, for PK could not be expressed in this strain, the difference existing in the two strains was pyk gene and adk1 gene exchanged the relevant location on plasmid pET-17b.
The possible reasons which would affect PK expression efficiency in strain E. coli BL 21(pET-adk1-pyk) were analyzed, the transcription level, mRNA stability and the gene-dependent sequence were not the crucial reasons responsible for PK translation efficiency, though they were thought important factors on heterogenous gene expression efficiency in the engineering bacteria [7, 8].
The 5’-UTR +25 bp fragment of the upstream and the gene 5’-terminal sequence of the first 11 codons from the start codon ATG was important on gene translation
 (a) (b)
(a) (b)
Figure 6. Predicted Secondary structure of pyk mRNA 5’-end the first 11 codons and the fragment of 5’UTR + 25 bp, totally the 58 nucleotides in BL21 (pET-adk1-pyk) and BL21 (pET-pyk-adk1) as (a) and (b).
Table 1. Comparing Free energy of the anlysis pyk mRNA secondary structure sequence in two recombinants.

initiation. The hairpin or loops formed in this region could afford the ribosomes different binding efficiency, and affected the formation of the initiation complex [4, 9]. pyk mRNA in the E. coli BL21 (pET-adk1-pyk) stain formed more hairpin structures than it were in E. coli BL21 (pET-pyk-adk1) predicted using the software MFOLD [7]. The calculated Gibbs free energy values (ΔG) of this region was –26.7 kJ·mol–1 in E. coli BL21 (pET-adk1-pyk) and –18.0 kJ·mol–1 E. coli BL21 (pETpyk-adk1), there were 31.8% higher energy barriers in E. coli BL21 (pET-pyk-adk1) than in E. coli BL21 (pETadk1-pyk). Zuker has reported that the –25.1 kJ·mol–1 Minimum Gibbs free energy of a gene mRNA secondary structure was the “on or off” switch for gene translation initiation [6]. The higher energy barrier of pyk mRNA in E. coli BL21(pET-adk1-pyk) will afford more energy barrier for initiating the protein translation as Pfleger [9] reported, and thus blocked the translation efficiency of pyk in this strain. However, it was not happening in E. coli BL21 (pET-pyk-adk1), both PK and AK could be expressed efficiently and used to catalyze dAMP to dATP.
Usually, a stronger mRNA secondary structure would enhance its stability and prolong the half-time of the mRNA. As for pyk mRNA in E. coli BL21 (pETadk1-pyk), the stronger secondary structure only inhibited its translation efficiency, have no effect on its halftime, the reasons might because of there have no endoribonuclease recognizing sequences in this region [10].
This study constructed a dATP conversion strain, and made it feasible to convert dATP from dAMP directly. The relative location has important effect on pyk gene translation efficiency. Only cloned pyk gene at an optimum location in the expression vector, the dATP conversion strain could be constructed. The recombinants could be used directly to convert 500 mM dAMP into dATP, after a simple purification, above 95% purity dATP could be produced, and the yield was above 89%, it was much higher than the chemical method used recently.
5. CONCLUSIONS
The gene location has very important effects on for pyk gene expression level in pET-17b plasmid. The 5’-UTR sequence including the upstream +25 bp fragment, and the first 11 codons from the start codon ATG are sensitive factors on PK translation initiation. Only the pyk gene was cloned ahead of adk1, the adenylate kinase (AK) and pyruvate kinase (PK) can be co-expressed in one recombinant strain and used directly to complete the two-step phosphorylation reaction, 500 mM dAMP substrates can be converted into dATP directly by one cell.
Using this simple, economically and environmentally friendly method the dATP yield was above 89%.
6. ACKNOWLEDGEMENTS
This research was supported by Science and Technology Commission of Shanghai Municipality (07ZR14031), Open Project for the State Key Laboratory of Bioreactor Engineering (2060204) and Fundamental Research Funds for the Central Universities of China (WF1114045).
REFERENCES
- Bao, J. and Ryu, D.D.Y. (2005) Biosynthesis reaction mechanism and kinetics of deoxynucleoside triphosphates dATP and dGTP. Biotechnology Bioengeering, 89, 485- 491. doi:10.1002/bit.20380
- Smolke, C.D. and Keasling, J.D. (2002) Effect of gene location, mrna secondary structures, and rnase sites on expression of two genes in an engineered operon. Biotechnology Bioengeering, 80, 762-776. doi:10.1002/bit.10434
- Alifano, P., Bruni, C.B. and Carlomagno, M.S. (1994) Control of mRNA processing and decay in prokaryotes. Genetica, 94, 157-172. doi:10.1007/BF01443430
- Dieter, V., Manfred, W., Cordula, N., Sabine, W. and Bernd, B. (2004) Analyzing and enhancing mRNA translational efficiency in an Escherichia coli in vitro expression system. Biochemical and Biophysical Research Communications, 318, 601-614. doi:10.1016/j.bbrc.2004.04.064
- Ravanshad, M., Sabahi, F. and Mahboudi, F. (2007) Quantification analysis of dot blot assays for human immunodeficiency virus type 1 and 2 antibodies. Iranian Journal of Basic Medical, 10, 132-138.
- Zuker, M. (2003) Web server for nucleic acid folding and hybridization prediction. Nucleic Acids Research, 31, 3406-3415. doi:10.1093/nar/gkg595
- Kimura, S. and Iyanagi, T. (2003) High-level expression of porcine liver cytochrome P-450 reductase catalytic domain in Escherichia coli by modulating the predicted local secondary structure of mRNA. Journal of Biochemistry, 134, 403-413.
- Geissmann, T., Marzi, S. and Romby, P. (2009) The role of mRNA structure in translational control in bacteria. RNA Biology, 6, 153-160. doi:10.4161/rna.6.2.8047
- Pfleger, B.F., Fawzi, N.J. and Keasling, J.D. (2005) Optimization of DsRed production in Escherichia coli: Effect of ribosome binding site sequestration on translation efficiency. Biotechnology Bioengeering, 92, 553-558. doi:10.1002/bit.20630
- Nilsson, G., Belasco, J.G., Cohen, S. N. and Von Gabain, A. (1987) Effect of premature termination of translation on messenger RNA stability depends on the site of ribosome release. Proceedings of the National Academy of Science, 84, 4890-4894. doi:10.1073/pnas.84.14.4890
NOTES
*Corresponding author.