Open Journal of Biophysics
Vol. 2 No. 4 (2012) , Article ID: 24099 , 4 pages DOI:10.4236/ojbiphy.2012.24014
Solid-State NMR Spectroscopic Approaches to Investigate Dynamics, Secondary Structure and Topology of Membrane Proteins
Department of Chemistry and Biochemistry, Miami University, Oxford, USA
Email: *abubaksn@muohio.edu
Received June 15, 2012; revised July 29, 2012; accepted August 20, 2012
Keywords: Solid-State NMR; Structure and Dynamics; Membrane Proteins
ABSTRACT
Solid-state NMR spectroscopy is routinely used to determine the structural and dynamic properties of both membrane proteins and peptides in phospholipid bilayers [1-26]. From the perspective of the perpetuated lipids, 2H solid-state NMR spectroscopy can be used to probe the effect of embedded proteins on the order and dynamics of the acyl chains of phospholipid bilayers [8-13]. Moreover, 31P solid-state NMR spectroscopy can be used to investigate the interaction of peptides, proteins and drugs with phospholipid head groups [11-14]. The secondary structure of 13C=O site-specific isotopically labeled peptides or proteins inserted into lipid bilayers can be probed utilizing 13C CPMAS solid-state NMR spectroscopy [15-18]. Also, solid-state NMR spectroscopic studies can be utilized to ascertain pertinent information on the backbone and side-chain dynamics of 2Hand 15N-labeled proteins, respectively, in phospholipid bilayers [19-26]. Finally, specific 15N-labeled amide sites on a protein embedded inside oriented bilayers can be used to probe the alignment of the helices with respect to the bilayer normal [2]. A brief summary of all these solid-state NMR approaches are provided in this minireview.
1. Introduction
Membrane proteins make up approximately one-third of the total number of known proteins [27]. They play several significant roles in biological systems that includes transporting ions, acting as receptors, participating in membrane fusion and destabilization, and many others. Despite the abundance and clear importance of membrane-associated proteins, limited information about these systems exists. Structural studies of these membrane proteins are key to understand their biological functions. X-ray crystallography has been used to elucidate structural information of biologically significant protein systems [28-35]. However, the hydrophobic surfaces associated with membrane-bound protein systems make the crystallization process very challenging. Although investigators are making progress with X-ray techniques, still only a limited list of membrane protein structures have been obtain via X-ray crystallography [36]. Alternatively, solid-state NMR spectroscopy is a powerful technique that can be used to provide structural, orientational, and dynamic information about membrane protein systems in model membrane systems [2,37,38].
2. Solid-State NMR Approaches
2.1. 2H Solid-State NMR Spectroscopy of Lipids with 2H-Lableled Acyl Chains
Multilamellar vesicles (MLVs) can be prepared from phospholipids such as 1-palmitoyl-2-oleoyl-sn-glycero-phosphocholine (POPC) and the details of sample preparation and type of lipid was reported previously [3-7]. Approximately 10% of deuterated POPC (sn-1 chain, POPC-d31, see Figure 1) or any other deuterated lipid of interest can be added to each sample. Other lipids can be used such as the negatively charged lipids such as 1-palmitoyl(d31)-2-oleoyl-sn-glycero-3-[phospho-L-serine] phospholipids (POPS-d31) used in Sap C studies (see Figure 2" target="_self"> Figure 2) and Distearoyl-d70-phosphatidylglycerol (DSPGd70) (see Figure 3).
The effect of embedded proteins on the order and dynamics of the acyl chains of POPC-d31 bilayers can be investigated utilizing 2H solid-state NMR spectroscopy in the absence as well as in the presence of (X) mol% of the protein with respect to the lipids at different temperatures [40]. For example, in Figure 4(a), the central resonance doublet corresponds to the terminal CD3 groups (Carbon # 1 in Figure 1) and the remaining over-lapped doublets result from the different CD2 segments of the acyl chain of POPC-d31. The addition of (X) mol% of specific protein with respect to the lipids to POPC MLVs could alter (depending on the protein of interest) the lineshape and the spectral resolution of the 2H NMR spectra (Figure 4(b)) when compared to the control (Figure 4(a)) sample.
In Figure 4(b), the loss in spectral resolution is manifested by the disappearance of sharp edges of the 2H NMR peaks. The changes in spectral resolution of the 2H NMR spectra indicate that the protein interacts with the POPC-d31-containing MLVs when compared to the control. If the spectral resolution and the line shapes did not change when compared to the control, this indicates that the protein does not interact significantly with the POPC MLVs as indicated in a previous study from the Lorigan Lab using the transmembrane domain segment of WTPLB [12].
All the powder-type 2H NMR spectra of multilamellar dispersions of POPC-d31 can be numerically deconvoluted (dePaked) using the algorithm of McCabe and Wassal [41,42]. These spectra can be dePaked such that the bilayer normal was perpendicular with respect to the direction of the static magnetic field. Then, the quadrupolar splittings can be directly measured from the dePaked spectra and converted into order parameters as described before [11,12]. The quadrupolar splittings of the CD3 methyl groups (see Figure 1) at the end of the acyl chains are the smallest and close to 0 kHz because
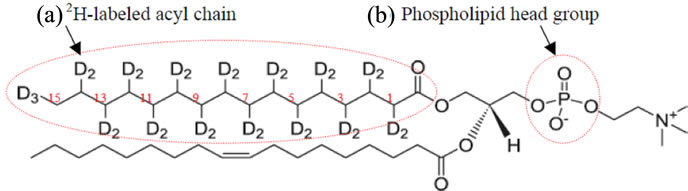
Figure 1. POPC-d31 structure [39]. The 2H-labeled acyl chain and phospholipids head groups are labeled with dashed red circles. The numbering of the 2H-labeled carbon skeleton of the acyl chain is shown is red.
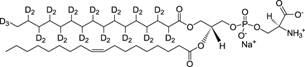
Figure 2. POPS-d31 structure [39]. Only one acyl chain is 2H-labeled.

Figure 3. Distearoyl-d70-phosphatidylglycerol (DSPG-d70) structure [39].
they rotate at the fastest frequency. The second smallest splitting was assigned to the 2H attached to C-14 and so forth along the acyl chain. The quadrupolar splittings for the deuterons in the plateau region were estimated by integration of the last broad peak.
The quadrupolar splittings can be directly measured from the dePaked spectra and converted into the SCD order parameter using the following expression;  = 3/4(e2qQ/h)
= 3/4(e2qQ/h)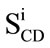 [43-45]. Where
[43-45]. Where  is the quadrupolar splitting for a deuteron attached to the ith carbon, e2qQ/h is the quadrupolar splitting constant (168 kHz for deuterons in C-2H bonds), and
is the quadrupolar splitting for a deuteron attached to the ith carbon, e2qQ/h is the quadrupolar splitting constant (168 kHz for deuterons in C-2H bonds), and  is the chain order parameter for a deuteron attached to the ith carbon of the acyl chain of POPC-d31. The order parameters calculated for the CD3 quadrupolar splitting should be multiplied by three according to procedures in the literature [46,47].
is the chain order parameter for a deuteron attached to the ith carbon of the acyl chain of POPC-d31. The order parameters calculated for the CD3 quadrupolar splitting should be multiplied by three according to procedures in the literature [46,47].
Figure 5 displays the smoothed segmental C-D bond order parameters (SCD) of the POPC-d31 acyl chains calculated by dePakeing the corresponding 2H NMR powder spectra shown in Figure 4. Figure 5 reveals a characteristic profile of decreasing order (SCD) with increasing distance from the glycerol backbone of POPC-d31 MLVs. Any decrease (if any) in the order parameter profile of POPC-d31-containing bilayers in the presence of (X) mol% of specific protein indicates more disorder and motion in the acyl chain region of the lipids when compared to the control sample (without protein). If the order parameter profile of POPC-d31-containing bilayers in the presence of (X) mol% protein is similar to the control sample, this indicates that this particular protein is not altering the order and motion in the acyl chain region of the lipids.
 (a)
(a) (b)
(b)
Figure 4. 2H NMR powder-pattern spectra of POPC-d31 bilayers in the absence (a) and in the presence of (X) mol% protein with respect to the lipids.

Figure 5. The smoothed acyl chains orientational order parameter (SCD) profiles calculated from the dePaked 2H NMR spectra of POPC-d31 (Figure 4). The closed squares and circles represent POPC bilayers with 0 mol% and (X) mol% protein, respectively.
2.2. 31P Solid-State NMR Spectroscopy of Phospholipid Head Groups
The motionally averaged powder pattern spectra are characteristic of MLVs in the liquid crystalline phase (La) and are expected for phospholipids bilayers at a temperature well above the chain melting point transition temperature (Tm) [48]. Figure 6(a) shows the structure of the neutral phospholipid POPC (Tm = –2˚C) [39]. In addition, other phospholipids that includes 1-palmitoyl-2- oleoyl-sn-glycero-3-[phospho-L-serine] (POPS, Tm = 14 ˚C, Figure 6(b)), dioleoylphosphatidyl-glycerol (DOPG, Tm = –18˚C, Figure 6(c)) and dioleoylphosphatidylserine (DOPS, Tm = –11˚C, Figure 6(d)) structures are shown [39]. The static 31P NMR spectrum of each phospholipid head group shown in Figures 6(a)-(d) have a specific 31P chemical shift anisotropy (CSA; is equal s33-s11) width and lineshape. In several cases, the lineshapes of the static 31P NMR spectra and its corresponding CSA values have been successfully used to study the perturbation effect induced by drugs and proteins on phospholipids [11,14,49,50]. The addition of (X) mol% of specific protein to specific MLVs could alter the 31P CSA and line shape (Figure 7(b)) when compared to the control (Figure 7(a)) sample. This indicates that the protein of interest is interacting with the head group region of the MLVs.
2.3. 13C Solid-State NMR Spectroscopy of Site-Specific 13C-Labeled Peptides in MLVs
The secondary structure of 13C=O site-specific isotopically labeled peptides or proteins inserted into lipid bilayers can be probed utilizing 13C CPMAS solid-state NMR spectroscopy [15-18,50]. The local conformations of peptides and proteins can be characterized by examining the 13C=O chemical shifts of the 13C-labeled carbonyl of Ala, Leu, and Val. In general, the a-helical structure chemical shifts may vary within the data range (174 - 177 ppm) for different conformational-dependent changes (see Figure 8(a)) [17]. Also, a single 13C NMR peak at approximately 172.4 ppm can be attributed to either a b-sheet or an unstructured structural conformation of the peptide (see Figure 8(b)); whereas a single peak at approximately 176 ppm can be attributed to one a-helical structure conformation (see Figure 8(c)) [15].

Figure 6. The structure of (a) POPC, (b) POPS, (c) DOPG and (d) DOPS phospholipids [39].
 (a)
(a)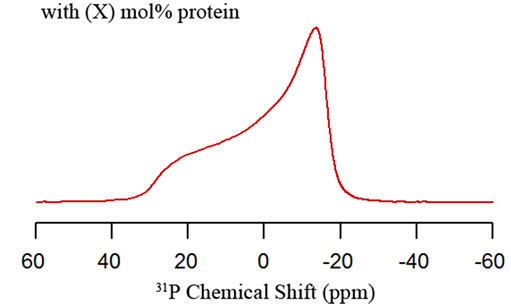 (b)
(b)
Figure 7. Static 31P solid-state NMR spectra of phospholipid MLVs in the absence (a) and in the presence (b) of X mol% protein.

Figure 8. 13C=O chemical shifts of 13C-labeled carbonyl of Ala located at (A) α-helix with two structural conformations, (B) Unstructured or β-sheet structural conformation and (C) α-helical with one structure conformation.
2.4. 2H Solid-State NMR Spectroscopy of Site-Specific 2H-Labeled Peptides in MLVs
As mentioned previously, probing of how both segments of phosphorylated and unphosphorylated membrane proteins are moving within the phospholipid bilayers is crucial to describe the physiological functions. 2H solidstate NMR spectroscopy is a powerful well developed technique to study the structural and side-chain dynamic properties of membrane proteins in phospholipid bilayers [8,51-53]. The corresponding quadrupolar splitting and lineshapes of the 2H solid-state NMR spectra can be used to probe the molecular dynamics of the sidechain of selectively labeled residues in site-specific 2H-labeled integral membrane proteins [19-24].
In previous studies, methyl group motions have been well-characterized utilizing 2H NMR studies of CD3-labeled sites of alanines, valines and leucines [19,24,25,54-57]. For the isotopically labeled alanines (short aliphatic side-chains), the deuterated methyl group (CD3) rotates along the Ca-Cb bond and allows the deuterons to make jumps between three-sites described by a tertrahedral geometry (see Figure 9(a)) [58,59]. However, for Leu, the long aliphatic side-chain can be isotopically labeled at the δ- and/or ε-CD3 sites (see Figure 9 (b)) and the deuterium NMR powder pattern lineshapes will be strongly influenced by the motions about the Cg-Cd bond axis as well as by additional librational motion about the Ca-Cb and Cb-Cg bond axes at various temperatures [24,60].
It has been reported that if the CD3-methyl probe of a protein undergoes no motion other than those associated with the axial rotation about the C-CD3 bond in a randomly dispersed sample, the resultant 2H NMR spectra will consist of a Pake pattern with a 40 kHz quadrupolar splitting (see Figure 10(a)) [61]. However, residues located outside the membrane are expected to be more motionally averaged and yield an isotropic peak (see Figure 10(b)) [62].
2.5. 15N Solid-State NMR Spectroscopy of Site Specific 15N-Labeled Peptides in MLVs
15N solid-state NMR spectroscopic studies are utilized to ascertain pertinent information on the backbone structure and dynamics of membrane proteins in phospholipid bilayers [25,26]. It provides important physiological and mechanistic information regarding the regulatory role of membrane proteins and it phosphorylated form in biological systems [64-66]. Generally, the immobile (without large amplitude motions) amide sites of specific 15N-labeled proteins yield a broad static 15N powder pattern (see Figure 11(a)); whereas, motionally averaged amide sites reveal isotropic peaks (see Figure 11(b)) [26].

Figure 9. The structure of (a) alanine and (b) leucine residues.
 (a)
(a) (b)
(b)
Figure 10. 2H NMR simulations of CD3-Ala using Multiple Axis Quadrupolar Echo Tailing (MXQET) program [63]. The model simulations show (a) Pake pattern and (b) isotropic peak spectra.
 (a)
(a)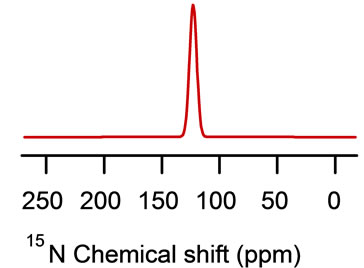 (b)
(b)
Figure 11. Simulations of the 15N NMR spectra using the Dominique Massiot’s Fit (DMFIT) software program [67]. The model simulations show (a) a powder pattern and (b) an isotropic peak.
2.6. 15N Solid-State NMR Spectroscopy of Site-Specific 15N-Labeled Peptides in Mechanically Oriented Bilayers
Determining the structural topology of membrane proteins and its interaction with the lipids is critical to understand its physiological regulatory function. 15N solidstate NMR spectroscopy is a powerful tool to ascertain direct information regarding the structural topology of membrane proteins in oriented phospholipid bilayers [2].
In this approach, specific 15N labeled amide sites can be used to probe the alignment of the helix with respect to the bilayer normal. For example, this technique was used to probe the orientation of both the transmembrane and cytoplasmic domains of WT-PLB embedded inside mechanically oriented phospholipids [68]. Site-specific 15N-labeled WT-PLB were chosen at Ala11 (in the cytoplasmic domain) and Leu 42 and Leu 51 (both in the transmembrane helix) (see Figure 12).
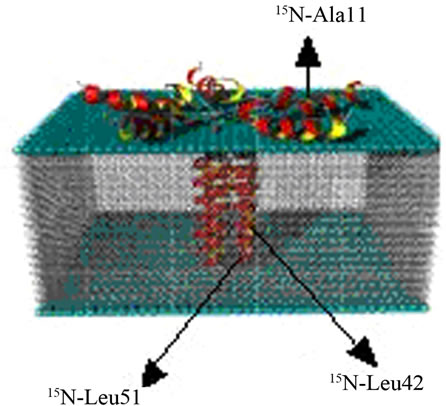
Figure 12. Side-view of a proposed structural model of site-specific 15N-labeled WT-PLB at Ala11 (in the cytoplasmic domain) and Leu 42 and Leu 51 (both in the transmembrane helix) in oriented bilayers.
A resonance peak at approximately 70 ppm (close to the s^ component of the chemical shift tensor of the corresponding powder spectrum, see Figure 13), indicates that this residue is oriented approximately perpendicular to the bilayer normal (transmembrane helix as shown in Figure 12) [2]. Also, a 15N resonance peak at approximately 210 ppm (close to the sêê component of the chemical shift tensor of the corresponding powder spectra, see Figure 13) indicates that the amide backbone vector of this residue is nearly parallel to the bilayer normal (the helix lies on the surface of the phospholipids bilayers as shown for cytoplasmic domain of the proposed structural model of WT-PLB presented in Figure 12 [2].
3. Conclusion
Solid-state NMR spectroscopy is routinely used to determine the structural and dynamic properties of both membrane proteins and peptides in phospholipid bilayers. Together, 2H, 31P, 13C and 15N solid-state NMR spectroscopy can be used to probe the effect of embedded proteins on the order and dynamics of the acyl chains, phospholipid head group as well as the secondary structure of site-specific isotopically labeled amino acid and helix orientation with respect to the membrane. A summary of all these solid-state NMR approaches are provided in this mini-review.
 (a)
(a) (b)
(b)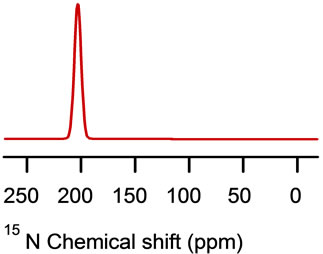 (c)
(c)
Figure 13. Simulations of the 15N NMR spectra using the DMFIT software program [67]. The model simulations show (a) powder pattern and (b) 15N resonance peak at approximately 70 ppm (close to the σ^ component of the chemical shift tensor of the corresponding powder spectrum) and (c) 15N resonance peak at approximately 210 ppm (close to the σêê component).
REFERENCES
- T. A. Cross, “Solid-State Nuclear Magnetic Resonance Characterization of Gramicidin Channel Structure,” Methods in Enzymology, Vol. 289, 1997, pp. 672-696.
- T. A. Cross and S. J. Opella, “Solid-State NMR Structural Studies of peptides and Proteins in Membranes,” Current Opinion in Structural Biology, Vol. 4, No. 4, 1994, pp. 574-581. doi:10.1016/S0959-440X(94)90220-8
- J. R. Long, W. J. Shaw, P. S. Stayton and G. P. Drobny, “Structure and Dynamics of Hydrated Staherin on Hydroxyapatite as Determined by Solid-State NMR,” Biochemistry, Vol. 40, No. 51, 2001, pp. 15451-15455.
- R. Mani, J. J. Buffy, A. J. Waring, R. I. Lehrer and M. Hong, “Solid-State NMR Investigation of the Selective Disruption of Lipid Membranes by Protegrin-1,” Biochemistry, Vol. 43, No. 43, 2004, pp. 13839-13848. doi:10.1021/bi048650t
- I. Marcotte, E. J. Dufourc, M. Ouellet and M. Auger, “Interaction of the Neuropeptide Met-Enkephalin with Zwitterionic and Negatively Charged Bicelles as Viewed by 31P and 2H Solid-State NMR,” Biophysical Journal, Vol. 85, No. 1, 2003, pp. 328-339.
- Y. Nakazawa and T. Asakura, “Structure Determination of a Peptide Model of the Repeated Helical Dmoain in Samia Cynthia Ricini Silk Fibroin before Spinning by a Combination of Advanced Solid-State NMR Methods,” Journal of the American Chemical Society, Vol. 125, No. 24, 2003, pp. 7230-7237.
- A. Watts, “Solid-State NMR Approaches for Studying the Interaction of Peptides and Proteins with Membranes,” Biochim Biophys Acta, Vol. 37, No. 2, 1998, pp. 297-318.
- B. W. Koenig, J. A. Ferritti and K. Gawrisch, “Site-Specific Deuterium Order Parameters and Membrane-Bound Behavior of a Peptide Fragment from the Intracellular Domain of HIV-1 gp-41,” Biochemistry, Vol. 38, No. 19, 1999, pp. 6327-6334.
- G. Laroche, E. J. Dufourc, M. Pezolet and J. Dufourcq, “Coupled Changes between Lipid Order and Polypeptide Conformation at the Membrane-Surface: A 2H NMR and Raman-Study of Polylysine Phosphatidic-Acid Systems,” Biochemistry, Vol. 29, No. 27, 1990, pp. 6460-6465. doi:10.1021/bi00479a018
- S. Yamaguchi, D. Huster, A. Waring, R. I. Lehrer, W. Kearney, B. F. Tack and M. Hong, “Orientation and Dynamics of an antimicrobial Peptide in the Lipid Bilayer by Solid-State NMR Spectroscopy,” Biophysical Journal, Vol. 81, No. 4, 2001, pp. 2203-2214. doi:10.1016/S0006-3495(01)75868-7
- S. Abu-Baker, X. Qi, J. Newstadt and G. A. Lorigan, “Structural Changes in a Binary Mixed Phospholipid Bilayer of DOPG and DOPS upon Sap C Interaction at Acidic pH Utilizing 31P and 2H Solid-State NMR Spectroscopy,” Biochimica et Biophysica Acta, Vol. 17, No. 1, 2005, pp. 58-66.
- P. C. Dave, E. K. Tiburu, K. Damodaran and G. A. Lorigan, “31P and 2H Solid-State NMR Spectroscopic Studies of the Transmembrane Domain of the Membrane-Bound Protein Phospholamban,” Biophysical Journal, Vol. 86, 2004, pp. 1564-1573.
- J. Lu, K. Damodaran, J. Blazyk and G. A. Lorigan, “Solid-State Nuclear Magnetic Resonance Relaxation Studies of the Interaction Mechanism of Antimicrobial Peptides with Phospholipid Bilayer Membranes,” Biochemistry, Vol. 44, No. 30, 2005, pp. 10208-10217.
- J. S. Santos, D. K. Lee and A. Ramamoorthy, “Effects of Antidepressants on the Conformation of Phospholipid Headgroups Studied by Solid-State NMR,” Magnetic Resonance in Chemistry, Vol. 42, No. 2, 2004, pp. 105-114.
- D. J. Hirsh, J. Hammer, W. L. Maloy, J. Blazyk and J. Schaefer, “Secondary Structure and Location of a Magainin Analogue in Synthetic Phospholipid Bilayers,” Biochemistry, Vol. 35, No. 39, 1996, pp. 12733-12741. doi:10.1021/bi961468a
- S. Kimura, A. Naito, S. Tuzi and H. Saito, “Dynamics and Orietation of Transmembrane Peptide from Bacteriorhodopsin Incorporated into Lipid Bilayer as Revealed by Solid State 31P and 13C NMR Spectroscopy,” Biopolymers, Vol. 63, No. 2, 2002, pp. 122-131.
- S. Tuzi, A. Naito and H. Saito, “A High-Resolusion SolidState 13C-NMR Study on [1-13C]Ala and [3-13C] Ala and [1-13C]Leu and Val-Labelled Bacteriorhodopsin: Conformation and Dynamics of Transmembrane Helics, Loops and Termini, and Hydration-Induced Conformational Changes,” European Journal of Biochemistry, Vol. 218, No. 3, 1993, pp. 837-844.
- S. Yamaguchi, K. Shimono, Y. Sudo, S. Tuzi, A. Naito and N. Kamo, “Conformation and Dynamics of the [3- 13C]Ala, [1-13C]Val-Labeled Truncated Pharaonis Transducer, pHtrII(1-159), as Revealed by Site-Directed 13C Solid-State NMR: Changes Due to Association with Phoborhodopsin (Sensory Rhodopsin II),” Biophysical Journal, Vol. 86, No. 5, 2004, pp. 3131-3140.
- L. S. Batchelder, C. E. Sullivan, L. W. Jelinski and D. A. Trochia, “Characterization of Leucine Side-Chain Reorientation in Collagen-Fibrils by Solid-State Deuterium NMR,” Proceedings of the National Academy of Sciences of the USA, Vol. 79, No. 2, 1982, pp. 386-389.
- K. Beshah, E. T. Olejniczak and R. G. Griffin, “Deuterium NMR Study of Methyl Dynamics in L-Alanine,” Journal of Chemical Physics, Vol. 86, No. 9, 1987, pp. 4730-4736.
- M. A. Keniry, A. Kintanar, R. L. Smith, H. S. Gutowsky and E. Oldfield, “Nuclear Magnetic Resonance Relaxation of Deuteriomethyl-Labeled Amino Acids Crystals and in Halobacterium Halobium and E. coli Cell Membranes,” Biochemsitry, Vol. 23, 1984, pp. 288-298.
- R. A. Kinsey, A. Kintanar and E. Oldfield, “Dynamics of Amino Acid Side-Chains in Membrane Proteins by High Field Solid-State Deuterium Nuclear Magnetic Resonance Spectroscopy,” The Journal of Biological Chemistry, Vol. 256, No. 17, 1981, pp. 9028-9036.
- G. C. Leo, L. A. Colnago, K. G. Valentine and S. J. Opella, “Dynamics of fd Coat Protein in Lipid Bilayers,” Biochemistry, Vol. 26, No. 3, 1987, pp. 854-862. doi:10.1021/bi00377a029
- W. Ying, S. E. Irvine, R. A. Beekman, D. J. Siminovitch and S. O. Smith, “Deuterium NMR Reveals Helix Packing Interactions in Phospholamban,” Journal of the American Chemical Society, Vol. 122, No. 45, 2000, pp. 11125-11128.
- E. K. Tiburu, P. C. Dave, K. Damodaran and G. A. Lorigan, “Investigating Leucine Side-Chain Dynamics and Backbone Conformations of Phospholamban Incorporated in Phospholipid Bilayers Utilizing 2H and 15N SolidState NMR Spectroscopy,” Biochemistry, Vol. 43, No. 44, 2004, pp. 13899-13909. doi:10.1021/bi0490993
- L. A. Colnago, K. G. Valentine and S. J. Opella, “Dynamics of fd Coat Protein in Bacteriophage,” Biochemistry, Vol. 26, No. 5, 1987, pp. 847-854.
- W. L. Smith, R. M. Garavito and S. Fergusson-Miller, “Membrane Protein Structural Biology Minireview Series,” The Journal of Biological Chemistry, Vol. 276, No. 35, 2001, pp. 32393-32394. doi:10.1074/jbc.R100044200
- D. A. Doyle, J. M. Cabral, R. A. Pfuetzner, A. L. Kuo, J. M. Gulbis, S. L. Cohen, B. T. Chait and R. MacKinnon, “The Structure of the Potassium Channel: Molecular Basis of K+ Conduction and Selectivity,” Science, Vol. 280, No. 5360, 1998, pp. 69-77. doi:10.1126/science.280.5360.69
- G. Chang, R. H. Spencer, A. T. Lee, M. T. Barclay and D. C. Rees, “Structure of the MscL Homolog from Mycobacterium Tuberculosis: A Gated Mechanosensitive Ion Channel,” Science, Vol. 282, No. 5397, 1998, pp. 2220-2226. doi:10.1126/science.282.5397.2220
- Y. X. Jiang, A. Lee, J. Y. Chen, V. Ruta, M. Cadene, B. T. Chait and R. MacKinnon, “X-Ray Structure of a Voltage-Dependent K+ Channel,” Nature, Vol. 423, No. 6935, 2003, pp. 33-41. doi:10.1038/nature01580
- Y. X. Jiang, A. Lee, J. Y. Chen, M. Cadene, B. T. Chait, and R. MacKinnon, “Crystal Structure and Mechanism of a Calcium-Gated Potassium Channel,” Nature, Vol. 417, No. 6888, 2002, pp. 515-522. doi:10.1038/417515a
- Y. X. Jiang, A. Lee, J. Y. Chen, M. Cadene, B. T. Chait, and R. MacKinnon, “The Open Pore Conformation of Potassium Channels,” Nature, Vol. 417, No. 6888, 2002, pp. 523-526. doi:10.1038/417523a
- R. MacKinnon, “Nothing Automatic about Ion-Channel Structures,” Nature, Vol. 416, No. 6878, 2002, pp. 261-262. doi:10.1038/416261b
- A. Ben-Shem and D. Fass, “Structural Basis for Intramembrane Proteolysis by Rhomboid Serine Protease,” Proceedings of the National Academy of Sciences of the USA, Vol. 102, No. 2, 2007, pp. 401-402.
- M. J. Lemieux, S. J. Fischer, M. M. Cherney, M. M. Bateman and M. N. James, “The Crystal Structure of Rhomboid Peptidase from Haemophilus Influenzae Provides Insight into Intramembrane Proteolysis,” Proceedings of the National Academy of Sciences of the USA, Vol. 104, No. 3, 2007, pp. 750-754. doi:10.1073/pnas.0609981104
- Stephen White Laboratory Official Website, 2007. www.blanco.biomol.uci.edu/Membrane_proteins_xtal.html
- S. J. Opella and F. M. Marassi, “Structure Determination of Membrane Proteins by NMR Spectroscopy,” Chemical Reviews, Vol. 104, No. 8, 2004, pp. 3587-3606. doi:10.1021/cr0304121
- S. J. Opella, “NMR and Membrane Proteins,” Nature Structural & Molecular Biology, Vol. 4, 1997, pp. 845-848.
- Avanti Company Official Website, 2006. http://www.avantilipids.com/TechnicalInformation.html
- S. Abu-Baker and G. A. Lorigan, “Phospholamban and Its Phosphorylated form Interact Differently with Lipid Bilayers: A 31P, 2H, and 13C Solid-State NMR Spectroscopic Study,” Biochemistry, Vol. 45, No. 44, 2006, pp. 13312-13322.
- M. A. McCabe and S. R. Wassall, “Fast-Fourier-Transform Depaking,” Journal of Magnetic Resonance, Series B, Vol. 106, No. 1, 1995, pp. 80-82. doi:10.1006/jmrb.1995.1013
- M. A. McCabe and S. R. Wassall, “Rapid Deconvolution of NMR Powder Spectra by Weighted Fast Fourier Transformation,” Solid State Nuclear Magnetic Resonance, Vol 10, No. 1-2, 1997, pp. 53-61. doi:10.1016/S0926-2040(97)00024-6
- P. C. Dave E. K. Tiburu, K. Damodaran and G. A. Lorigan, “Investigating Structural Changes in the Lipid Bilayer upon Insertion of the Transmembrane Domain of the Membrane-Bound Protein Phospholamban Utilizing 31P and 2H Solid-State NMR Spectroscopy,” Biophysical Journal, Vol. 86, No. 3, 2004, pp. 1564-1573.
- P. C. Dave, E. K. Tiburu, N. A. Nusair and G. A. Lorigan, “Calculating Order Parameter Profiles Utilizing Magnetically Aligned Phospholipid Bilayers for 2H Solid-State NMR Studies,” Solid State Nuclear Magnetic Resonance, Vol. 24, No. 2-3, 2003, pp. 137-149. doi:10.1016/S0926-2040(03)00052-3
- D. Huster, X. Yao, K. Jakes and M. Hong, “Conformational Changes of Colicin Ia Channel-Forming Domain upon Membrane Binding: A Solid-State NMR Study,” Biochimica et Biophysica Acta, Vol. 1561, No. 2, 2002, pp. 159-170.
- D. Huster, K. Arnold and K. Garwrisch, “Influence of Docosahexaenoic Acid and Cholesterol on Lateral Lipid Organization,” Biochemistry, Vol. 37, No. 49, 1998, pp. 17299-17308.
- G. W. Stockson, C. F. Polnaszek, A. P. Tulloch, F. Hasan and I. C. P. Hasan, “Molecular Motion and Order in Single Bilayer Vesicles and Multilamellar Dispersions of Egg Lecithin and Lecithin-Cholesterol Moxtures. A Deuterium Nuclear Magnetic Resonance Study of Specifically Labeld Lipids,” Biochemistry, Vol. 15, 1976, pp. 954-966.
- J. Seelig, “31P Nuclear Magnetic Resonance and the Head Group Structure of Phospholipids in Membranes,” Biochimica et Biophysica Acta, Vol. 515, 1978, pp. 105-140.
- C. D. Pointer-Keenan, D. K. Lee, K. J. Hallock, A.Tan, R. Zand and A. Ramamoorthy, “Investigation of the Interaction of Myelin Basic Protein with phospholipid Bilayers Using Solid-State NMR Spectroscopy,” Chemistry and Physics of Lipids , Vol. 132, No. 1, 2004, pp. 47-54.
- S. Abu-Baker and G. A. Lorigan, “Phospholamban and Its Phosphorylated Form Interact Differently with Lipid Bilayers: A 31P, 2H, and 13C Solid-State NMR Spectroscopic Study,” Biochemistry, Vol. 45, No. 44, 2006, pp. 13312-13322. doi:10.1021/bi0614028
- J. W. Mack, D. A. Torchia and P. M. Steinert, “SolidState NMR Studies of the Dynamics and Structure of Mouse Keratin Intermediate Filaments,” Biochemsitry, Vol. 27, No. 15, 1988, pp. 5418-5426.
- R. S. Prosser, S. I. Daleman and J. H. Davis, “The Structure of an Integral Membrane Peptide: A Deuterium NMR-Study of Gramicidin,” Biophysical Journal, Vol. 66, No. 5, 1994, pp. 1415-1428. doi:10.1016/S0006-3495(94)80932-4
- S. Sharpe, K. R. Barber, C. W. M. Grant and M. R. Morrow, “Evidence of a Tendancy of Self-Association of the Transmembrane Domain of ErbB-2 in Fluid Phospholipid Bilayers,” Biochemistry, Vol. 41, 2002, pp. 2341-2352.
- D. H. Jones, A. C. Rigby, K. R. Barber and W. M. Grant, “Oligoerization of EGF Receptor Transmembrane Domain: A 2H NMR Study in Lipid Bilayers,” Biochemsitry, Vol. 36, No. 41, 1997, pp. 12616-12624.
- J. A. Killian, M. J. Taylor and R. E. Koeppe, “Orientation of Valine-1 Side-Chain of the Gramicidin Transmembrane Channel and Implications for Channel Functioning: A Deuterium NMR Study,” Biochemsitry, Vol. 31, No. 46, 1992, pp. 11283-11290.
- K. C. Lee, S. Huo and T. A. Cross, “Lipid-Peptide Interface: Valine Conformation and Dynamics in Gramicidin Channel in a Lipid Bilayer,” Biochemsitry, Vol. 34, 1995, pp. 857-867.
- P. C. A. Van der Wel, E. Strandberg, J. A. Killian and R. E. Koeppe, “Geometry and Intrinsic Tilt of a Trypto Phan-Anchored Transmembrane Alpha-Helix Determined by H-2 NMR,” Biophysical Journal, Vol. 83, No. 3, 2002, pp. 1479-1488. doi:10.1016/S0006-3495(02)73918-0
- L. S. Batchelder, C. H. Niu and D. A. Torchia, “Methyl Reorientation in Polycrystaline Amino Acids in Peptides,” Journal of the American Chemical Society, Vol. 105, No. 8, 1983, pp. 2228-2231.
- S. Abu-Baker, J. X. Lu, S. Chu, C. C. Brinn, C. A. Makaroff and G. A. Lorigan, “Side Chain and Backbone Dynamics of Phospholamban in Phospholipid Bilayers Utilizing 2H and 15N Solid-State NMR Spectroscopy,” Biochemistry , Vol. 46, No. 42, 2007, pp. 11695-11706. doi:10.1021/bi700749q
- J. Janin and S. Wodak, “Conformation of Amino Acid Side-Chain in Proteins,” Journal of Molecular Biology, Vol. 125, No. 3, 1978, pp. 357-386.
- A. C. Rigby, K. R. Barber, G. S. Shaw and C. W. M. Grant, “Transmembrane Region of the Epidermal Growth Factor Receptor: Behavior and Interactions via 2H NMR,” Biochemistry, Vol. 35, 1996, pp. 12591-12601.
- W. J. Gibbons Jr., E. S. Krap, N. A. Cellar, R. E. Minto and G. A. Lorigan, “Solid-State NMR Studies of a Diverged Microsomal Amino-Proximate Delta12 Desaturase Peptide Reveal Causes of Stability in Bilayer: Tyrosine Anchoring and Arginine Snorkeling,” Biophysical Journal, Vol. 90, No. 4, 2006, pp. 1249-1259.
- M. S. Greenfield, A. D. Ronemus, R. L. Vold, R. R. Vold, P. D. Ellis and T. E. Raidy, “Deuterium Quadrupole-Echo NMR Spectroscoppy III. Practice Aspects of line Shape Calculations for Multiaxis Rotational Processes,” Journal of Magnetic Resonance, Vol. 72, No. 1, 1987, pp. 89-107.
- K. Oxenoid and J. J. Chou, “The Structure of Phospholamban Pentamer Reveals a Channel-Like Architecture in Membrane,” Proceedings of the National Academy of Sciences of the USA, Vol. 102, No. 31, 2005, pp. 10870-10875. doi:10.1073/pnas.0504920102
- H. K. B. Simmerman and L. R. Jones, “Phospholamban: Protein Structure, Mechanism of Action, and Role in Cardiac Function,” Physiological Reviews, Vol. 78, No. 4, 1998, pp. 921-947.
- J. Zamoon, F. Nitu, C. Karim, D. D. Thomas and G. Veglia, “Mapping the Interaction Surface of a Membrane Protein: Unveiling the Conformational Switch of Phospholamban in Calcium Pump Regulation,” Proceedings of the National Academy of Sciences of the USA, Vol. 102, No. 13, 2005, pp. 4747-4752. doi:10.1073/pnas.0406039102
- D. Massiot, F. Fayon, M. Capron, I. King, S. Lecalve, B. Alonso, J. O. Durand, B. Bujoli, Z. Gan and G. Hoatson, “Modelling Oneand Two-Dimensional Solid-State NMR Spectra,” Magnetic Resonance in Chemistry, Vol. 40, No. 1, 2002, pp. 70-76.
- S. Abu-Baker, J. X. Lu, S. Chu, K. K. Shetty, P. L. Gor’kov and G. A. Lorigan, “The Structural Topology of WildType Phospholamban in Oriented Lipid Bilayers Using 15N Solid-State NMR Spectroscopy,” Protein Science, Vol. 16, No. 11, 2007, pp. 2345-2349.
NOTES
*Corresponding author.