Synthesis and Crystal Structure of the Hydrogen Bromide Salt of 1,4,7,10-Tetrakis(2-((4-methoxy)phenoxy) ethyl)-1,4,7,10-tetraazacyclododecane ()
1. Introduction
With the high abilities in recognizing specific DNA sequence and catalyzing hydrolysis of phosphate diester bonds, chemical nucleases have rapidly become an invaluable research tool in the fields of biology, bioorganic chemistry, therapy, and molecular biology [1] - [6] . Many complexes have been designed and studied. In this used ligands, cyclen with four benzyl groups, can coordinate with most mental cations or negative ion. The successful application of several cyclen derivative complexes for biomedical applications has stimulated interest for new cyclen-based ligands with different types of pendant arms in an attempt to find new ligands with different chemical, biological or catalytic properties [7] [8] [9] [10] [11] . Since armed cyclens have many structural and geometrical variations, they form a wide variety of metal complexes having specific sensing and signaling functions [12] .
Recently, many cyclen derivatives with different types of pendant arms have been designed and reported, but only a few compounds bearing four benzyl groups. Smith C. B. and coworkers reported the preparation of the cyclen derivatives: (R)-thppc12 (L) and (S)-thph-pc12 (L’), including four benzyl or phenoxy-methyl groups, where benzyl or phenoxymethyl groups project in the same direction due to the open nature of the electron-rich cavity [13] . If, in addition to bearing a donor atom, the pendant arms also have an aromatic moiety attached to them, the possibility arises of using the coordination of the ligand to a metal ion as a way of assembling a molecular receptor with a substantial cavity.
Encouraged by these considerations, we describe herein the synthesis of a new type of cyclen-base ligand with four neutral pendent groups at N residuals according to Scheme 1. Herein, X-ray crystal structure determination of compound 1 is undertaken to better understand the influence of structure modifications upon overall molecular geometry and conformation.
2. Experimental Section
2.1. Materials and Instruments
All chemicals were of reagent grade and used without further purification. All aqueous solutions were prepared from deionized or distilled water. Reaction and the resultant products were monitored by thin-layer chromatography (TLC) on Merck pre-coated silica gel F254 plates with separated compounds visualized at 254 nm under a UV lamp. Melting point (uncorrected) was determined on a XT4 MP: apparatus (Taike Corp, Beijing, China). NMR was recorded in CDCl3 on a Varian Mercury 300 spectrometer and resonance was given in ppm (δ) relative to TMS.
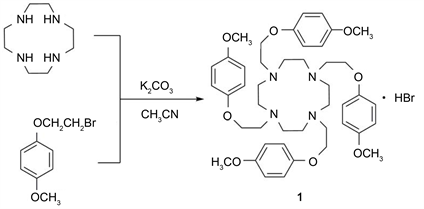
Scheme 1. Synthetic route to compound 1.
2.2. Synthesis of the Title Compound (1)
Compound 1 was synthesized based on literature method (Scheme 1) [14] . To a solution of cyclen (0.2 g, 1.16 mmol), 1-(2-bromoethoxy)-4-methoxybenzene (1.34 g, 5.8 mmol) in 30 mL CH3CN was added K2CO3 (0.83 g, 6.0 mmol). The resulting solution was heated under reflux and the progress of the reaction was monitored by TLC (CH2Cl2/CH3OH 7:1 silica gel). After disappearance of the starting material (ca. 8 h), the remaining solids were removed by filtration, and the filtrate was cooled to give compound 1, white solid, yield: 89.5%. Melting point: 128˚C - 129˚C. 1H NMR (300 MHz, CDCl3), δ (ppm): 6.79 - 6.69 (16H, dd, C6H4), 4.02 (8H, s, cyclen-CH2CH2O), 3.75 (12H, s, OCH3), 3.27 (8H, s, cyclen-CH2CH2O), 3.13 (16H, s, NCH2CH2N).
3. X-Ray Crystal Structure Determination
Crystallographic characteristics and X-ray-data collection and structure refinement parameters are presented in Table 1. Selected bond lengths are provided in Table 2. Selected bond angles are provided in Table S1. The crystal of 1 (0.2 mm ×
![]()
Table 1. Crystal data and structure refinement parameters of C44H61BrN4O8 (1).
![]()
Table 2. Selected bond angles for C44H61BrN4O8 (1).
Symmetry transformations used to generate equivalent atoms: −x + 1, y, −z + 3/2.
0.2 mm × 0.2 mm) was selected for data collection which was performed on a Bruker Apex II CCD diffractmeter equipped with a graphite-monochromatic CuKα radiation (λ = 0.71073 nm) at 293(2) K. A total of 24,759 reflections were collected in the range of 1.96 ≤ θ ≤ 28.08˚ (−22 ≤ h ≤ 22, −16 ≤ k ≤ 6 and −25 ≤ l ≤ 24), of which 24,759 were unique with Rint = 0.1233 and 5200 were observed with I > 2σ(I). Corrections for LP factors and semi-empirical absorption were applied based on Sadabs [15] . Unique data (Rint = 0.1233) were used to solve the structure by direct methods with SHELXS-97 program [16] . Most non-hydrogen atoms were determined with an E-map and the others were located in successive difference Fourier syntheses. Then, the final refinement was carried out by full-matrix least-squares methods with anisotropic thermal parameters for the non-hydrogen atoms on F2. The hydrogen atoms were located theoretically and refined with riding model position parameters and fixed isotropic thermal parameters. A full-matrix least-squares refinement gave the final R = 0.0434, wR = 0.1091 (w = 1/[σ2(Fo2) + (0.20P)2 + 0.00P], where P = (Fo2 + 2Fc2)/3), S = 0.89, (Δ/σ)max = 0.00, (Δρ)max = 0.624 and (Δρ)min = −0.498 e/Å3. The CIF file containing complete information on the studied structure was deposited with CCDC, deposition number 1,403,270, and is freely available upon request from the following web site: http://www.ccdc.cam.ac.uk/data_request/cif, and also available from the authors.
4. Results and Discussion
We describe herein the structure of the title compound 1 which is stable in air at room temperature. It is soluble in methanol and ethanol at higher temperature and soluble in CH2Cl2 and CHCl3. X-ray quality crystals of 1 were grown from ethanol by slow evaporation at room temperature for several days. The molecular structure of 1 crystallizes in orthorhombic space group Pbcn. Molecular structure of 1 is shown in Figure 1.
X-ray single-crystal structural analysis revealed that all the bond lengths and bond angles in the compound are within normal ranges and comparable to those corresponding in other similar compounds (Table 2 and Table S1). As shown in Figure 1, the asymmetric unit of 1 contains one half of organic cation, because it is situated on twofold axis. All the four hydroquinone groups of the benzene ring deviate in the same direction. They converge to form the resemblance of a binding π-electron-rich cavity. The depth of the cavity measured from the plane of O(1) and O(3) to the uppermost aromatic oxygen atom is 5.7 Å. The diameter of the cavity, defined as the shortest separation between the ring centroids of the three phenyl rings, is calculated to be 4.5 Å. There is no evidence for π・・・π interactions between the phenyl ring and another phenyl ring with their shortest separation larger than 4 Å. Similarly, Timothy S. R. et al. [17] also reported the defined boundaries of the π-electron-rich cavity via C-H・・・π stacking interactions, and commented on the cyclen derivatives, including four phenoxymethyl groups.
Hydrogen bonding interactions are usually important in the synthesis of supramolecular architecture [18] . In addition to the aromatic interactions, there are intermolecular C-H・・・O hydrogen bonding interactions between the C(9)-H(9A) and C(17)-H(17A) groups from benzyl groups carbon atoms (C(7) to C(12) and C(15) to C(20)) and O(2) and O(4) from methoxyl, resulting in an extended two-dimensional structure. But there are not intermolecular C-H・・・O hydrogen bonding interactions between (O(1) and O(3)) and benzyl groups carbon atoms (Figure 2). As shown in Figure 2, there are intramolecular C-H・・・Br hydrogen bonding interactions between the (C(2)-H(2A), and
![]()
Figure 1. Molecular structure of 1. All hydrogen atoms are omitted from the figure for clarity.
C(4)-H(4A)) from cyclen ring carbon atom (C(1) to C(8)) and (C(11)-H(11A) and C(21)-H(21A)) and Br(1). A three-dimensional structure is formed through these weak interactions among molecules. The molecules hydrogen bromides serve as the donor to carbon atoms, giving a total of crystallographically unique hydrogen bonds for 1.
The packing diagram is shown in Figure 3, where four molecules exist in the unit cell. The four molecules have a centrosymmetric distribution with the centroid of the unit cell. This assignment is favorable to crystal packing. There are two enantiomorphism crystals in the crystal packing diagram of 1. (A and D), (B and E) and (C and F) are mirror-related with each other (Figure 4).
![]()
Figure 2. Dimolecular graph of 1. All hydrogen atoms are omitted for clarity. Intramolecular C-H・・・Br and C-H・・・O hydrogen bonds are shown as dashed lines.
![]()
Figure 3. Crystal packing diagram of 1. Hydrogen atoms are omitted for clarity.
![]()
Figure 4. Crystal packing diagram of 1 in the unit cell along b.
5. Conclusion
In summary, this efficient one-step synthesis has yielded a novel molecule 1,4,7,10-tetrakis(2-((4-methoxy)phenoxy)ethyl)-1,4,7,10-tetraazacyclododecane 1. The cyclen derivatives contain four hydroquinone units carrying ethylene chains. All the four hydroquinone groups form a π-electron-rich cavity. This cavity-like structure of cyclen derivatives is expected to present interesting properties. Indeed, the reported cyclen derivatives bearing four hydroquinone moieties may play a significant role towards the improvement in metal coordination or small molecule induced by the reinforced cycles, due to the formation of the cavity-like structure. However, this work is poor in coordinating with metal ion or small molecule. These features are currently underway.
Acknowledgements
This research was supported financially by the National Natural Science Foundation of China (51506077), the Natural Science Foundation of Jiangsu Province (BK20150488), the Natural Science Foundation of Jiangsu High School (15KJB430007, 15KJB610003, 15KJD150002), the China Postdoctoral Foundation (2016M601733) and Research Foundation of Jiangsu University (13JDG066, 15JDG156).
Appendix
![]()
![]()
Table S1. Selected bond angles for C44H61BrN4O8 (1).