1. Introduction
Choroid plexus tumors are uncommon tumors which account for less than 1% of all brain tumors in adults and constitute approximately 1% to 5% of all childhood brain tumors based on different series [1] -[4] . They derive from choroid plexus epithelium and are characterized by papillary and intraventricular growth [4] . Choroid plexus tumors are categorized into choroids plexus papilloma (WHO grade I), choroid plexus carcinoma (WHO grade III), and atypical choroid plexus papilloma which is in-between on the basis of histological criteria [1] [4] -[6] . Choroid plexus papilloma accounts for 65% - 75% of the choroid plexus tumors [5] . Choroid plexus carcinoma is infrequent in adult patient; about 80% of choroid plexus carcinomas occur in pediatric population [4] . Because of the rarity of the choroid plexus carcinoma, the current treatment recommendations are based on little more evidence than expert opinions.
We report a rare case of choroid plexus carcinoma (WHO grade III) in the trigone and occipital horn of the left lateral ventricle in an adult patient who was successfully treated by a gross-total surgical resection followed by postoperative adjuvant radiotherapy and chemotherapy.
2. Case Report
In May 2011, a 21-year-old man with past medical history of asthma presented to us with the chief complaint of morning headache for one month. His headache was distent in nature and sometimes associated with nausea and vomiting. Before consulting us, he had visited a local hospital and had a brain computed tomography (CT) which showed a mass at the left occipital area. On this admission, the patient’s general physical examination and neurological examination were essentially normal. Preoperative complete blood count, screen panel, and chest x-ray film were within normal limits. Brain magnetic resonance imaging (MRI) was arranged which revealed a well-defined mass about 3.9 cm × 3.4 cm × 3.4 cm in size, either within the left lateral ventricle or extension from the brain parenchyma to the trigone and occipital horn of the left lateral ventricle. After Gadolinium administration, it showed heterogenous enhancement (Figure 1(A)). Under general anesthesia, the patient was placed in prone position and had undergone a left occipitoparietal craniotomy, posterior interhemispheric precuneus approach with the removal of the tumor. Under operating microscope, grossly, the tumor was red and soft (Figure 2(A)). At the late stage of tumor removal, the ventricle was entered and the choroid plexus inside the ventricle was seen (Figure 2(B)).
Histology examination showed that the specimen consisted papillary growth of fibrovascular connective tissue fronds covered by cuboidal to columnar epithelial cells. Focal increased cellular density, blurring of papillary pattern with sheets of tumor cells, numerous mitotic figures, focal necrosis, hemorrhage and nuclear atypia were evident. These tumor cells were immunoreactive to cytokeratin (AE1/AE3), but not to epithelial membrane antigen (EMA) stain. They also showed focal scattered positive in S100 and glial fibrillary acidic protein (GFAP) immunostains. Choroid plexus carcinoma (WHO grade III) was diagnosed based on the morphology of the tumor cells and the result of immunohistochemical stains (Figure 3).
Immediate postoperative imaging showed no obvious enhancing mass lesion in the left lateral ventricle (Figure 1(B)). Spine MRI in this admission and follow-up showed no evidence of abnormal signal intensity mass lesion in the spinal canal of the whole spine region.
The patient’s postoperative course was uneventful but his visual field study showed a right homonymous hemianopsia. To reduce the possibility of metastasis along the cerebrospinal fluid pathways and local recurrence, we administered postoperative adjuvant craniospinal irradiation as follows: craniospinal region 36 Gy × 20 fractions of total 720 Gy, tumor bed 54 Gy × 30 fractions of total 1620 Gy. Chemotherapy with DICE regimen (dexamethasone, ifosfamide, cisplatin, etoposide) after that irradiation was recommended by the oncologist. Totally, he received four cycles of postoperative adjuvant chemotherapy.
This patient is regularly followed up at our neurosurgical out-patient clinic. He is doing well and had back to college to continue his study in 2012. The last imaging follow-up in 2013 showed neither residual nor recurrent tumor (Figure 1(C)).
3. Discussion
Choroid plexus carcinoma (WHO grade III) is an uncommon, aggressive, malignant intracranial neoplasm that typically occurs in children [1] -[4] . Choroid plexus carcinomas arise from the choroid plexus epithelium [4] . The locations of the lesions, in general, are as follows: 50% of lateral ventricles, 40% of fourth ventricles, 5% of third ventricles, and 5% of multiple ventricular involvements [4] . They have been associated with obstruction of the cerebrospinal fluid pathways and, potentially, overproduction of cerebrospinal fluid, causing hydrocephalus and in-

Figure 1. Brain MRI, T1-weighted postcontrast image, axial view. A: preoperative study showed a heterogenous enhanced mass located either within the left lateral ventricle or extension from the brain parenchyma to trigone and occipital horn of the left lateral ventricle. (The red arrow pointed at the tumor). B: immediated postoperative study showed no obvious enhanced mass lesion. C: last follow-up study in 2013 showed neither residual nor recurrent tumor.

Figure 2. A: Gross appearance of tumor demonstrated hypervascularity. B: The ventricle was entered and choroid plexus was seen.
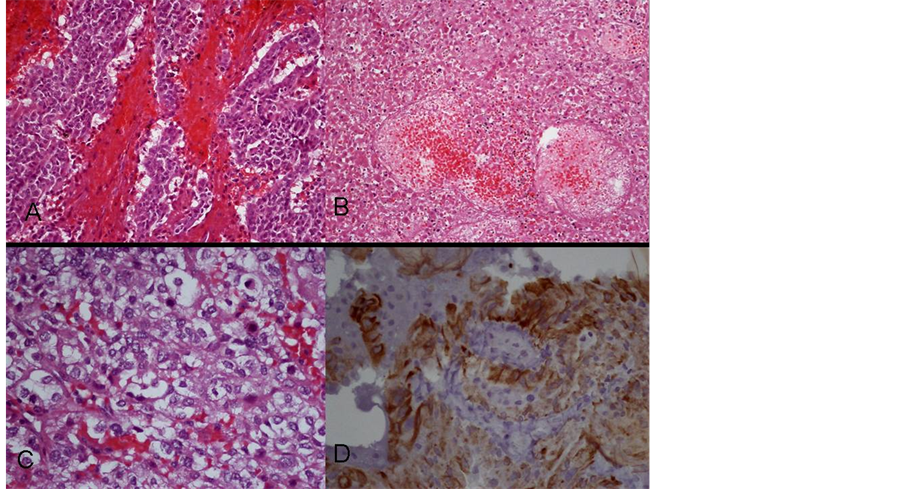
Figure 3. Histology of the tumor. A: H&E stain, 200×. Increased cellular density, blurring of papillary pattern with sheets of tumor cells were demonstrated. B: H&E stain, 200×. Tumor necrosis was evident. C: H&E stain, 400×. Numerous mitotic figures, nuclear atypia were displayed. D: Immunohistochemical staining showed positive to cytokeratin (AE1/AE3).
creased intracranial pressure [1] [4] . In infants, the clinical presentations include increased head circumference, delayed development, bulging fontanelles, separated sutures, strabismus or vomiting. In older children and adult patients, they may present with headache, vomiting, seizures, conscious disturbance, or neurological deficits [4] .
Histologically, as compared with choroid plexus papilloma (WHO grade I), choroid plexus carcinoma (WHO grade III) has the following characteristics: increased cellularity, increased mitotic figures (usually greater than 5 per 10 high-power fields), nuclear pleomorphism, necrosis, blurred papillary features. For immunohistochemical staining, choroid plexus carcinomas typically stain positive for cytokeratin, and demonstrate variable expression of vimentin, S100, transthyretin, and glial fibrillary acidic protein (GFAP). However, choroid plexus carcinoma typically negative for epithelial membrane antigen [4] .
Choroid plexus carcinoma is aggressive and is associated with dismal prognosis. The 5-year survival rates for choroid plexus carcinoma vary between 10% and 50% [1] -[6] . Currently, there is no established treatment protocol for choroid plexus carcinoma. Complete resection of this malignant tumor is the primary goal of treatment since it allows best chance of survival and improves the overall prognosis [1] [4] [5] . In cases of incomplete resection of choroid plexus carcinoma, second surgery may be helpful [4] -[6] . Postoperative radiotherapy is avoided in pediatric patients due to severe long-term sequelae, but it is considered in adult patients [3] [4] [6] . Adjuvant chemotherapy after surgery remains controversial [1] [2] [4] . Our patient presented with morning headache, nausea and vomiting which was a typical sign of increased intracranial pressure. He underwent a gross-total surgical resection of the tumor and received postoperative adjuvant radiation and chemotherapy. Until now, he has been disease-free for more than two years and has been back to school to continue his study. Choroid plexus carcinoma in adult is very rare and there is no established treatment protocol to this malignant tumor. We would like to share this successfully treated case with others to increase our clinical experience in the management of this infrequent malignant tumor.
NOTES
*Corresponding author.