iATC_Deep-mISF: A Multi-Label Classifier for Predicting the Classes of Anatomical Therapeutic Chemicals by Deep Learning ()
1. Introduction
According to the ATC (Anatomical Therapeutic Chemical) system (http://www.whocc.no/atc/structure_and_principles) as recommended by WHO (World Health Organization), the drug compounds are categorized into the following 14 main groups: 1) alimentary tract and metabolism; 2) blood and blood forming organs; 3) cardiovascular system; 4) dermatologicals; 5) genitourinary system and sex hormones; 6) systemic hormonal preparations, excluding sex hormones and insulins; 7) anti-infectives for systemic use; 8) antineoplastic and immunomodulating agents; 9) musculoskeletal system; 10) nervous system; 11) antiparasitic products, insecticides and repellents; 12) respiratory system; 13) sensory organs; 14) various. Given an uncharacterized compound, can we identify which ATC-class it belongs to? It is no doubt a significant problem for both basic research and drug development.
In 2017, a powerful predictor called “iATC-mISF”, was developed, which is overwhelmingly superior to its counterparts. But the method has not been further treated with the Deep Learning yet, a very powerful technique [1] [2]. The present study was devoted to doing so.
According to the 5-step guidelines [3] and demonstrated in a series of recent publications (see, e.g., [4] [5]), to develop a statistical predictor that not only can be easily used by experimental scientists but also can stimulate theoretical scientists to develop more relevant ones, we should make the following five steps crystal clear: 1) benchmark dataset, 2) sample formulation, 3) operation algorithm, 4) anticipated accuracy, and 5) web-server. Below, we are to elaborate how to deal with these procedures one-by-one.
2. Materials and Methods
2.1. Benchmark Dataset
The benchmark dataset used in this study is exactly the same as that in iATC-mSMF [6]; i.e.,
(1)
where the subset
only contains the samples from the m-th ATC class
and
denotes the symbol for “union” in the set theory. See Online Supporting Information S2 for a breakdown of the benchmark dataset according to the 14 subsets in Equation (1).
2.2. Installing Deep-Learning for Three Deeper Levels
In this study, we use multilayer perceptron neural network model, which consists of 3 fully connected layers and was used to predict classes of multi-label ATC classes, as illustrated in Figure 1. We set input layer with 14 neural unGranits which correspond to 14 features. Too many hidden layers would make network complexity bigger and suffer from the vanishing gradient problem while a model is constructed. Here, only two hidden layers are included. The hidden layer 1 is set as 200 neural units. The activation function is set as “relu”. The second hidden layer has 100 neural units. The activation function is set the same as the hidden layer 1. We end the model with 14 neural units and sigmoid activation. To go with it, we use the binary_crossentropy loss and the adam (adaptive moment estimation) optimizer to train the model. The metrics is set as “accuracy”. The batch size is set as 28, and the epochs is 100. The predicted results
![]()
Figure 1. An illustration to show a dense neural network with 3 fully connected layers. Adapted from [1] with permission.
were decided by the output of the threshold θ. If the output is greater than 0.5, the outcome was true; otherwise, false. For more information about this, see [1], where the details have been clearly elaborated and hence there is no need to repeat here.
The new predictor developed via the above procedures is called “iATC_Deep-mISF”, where “iATC_Deep” stands for “predict anatomical therapeutic chemicals”, and “mISF” for “multi-label classes”.
3. Results and Discussion
According to the 5-step rules [3], one of the important procedures in developing a new predictor is how to properly evaluate its anticipated accuracy. To deal with that, two issues need to be considered. 1) What metrics should be used to quantitatively reflect the predictor’s quality? 2) What test method should be applied to score the metrics?
3.1. A Set of Five Metrics for Multi-Label Systems
Different from the metrics used to measure the prediction quality of single-label systems, the metrics for the multi-label systems are much more complicated. To make them more intuitive and easier to understand for most experimental scientists, here we use the following intuitive Chou’s five metrics [7] or the “global metrics” that have recently been widely used for studying various multi-label systems (see, e.g., [8] [9]). For the current study, the set of global metrics can be formulated as:
(2)
where
is the total number of query proteins or tested proteins, M is the total number of different labels for the investigated system (for the current study it is
),
means the operator acting on the set therein to count the number of its elements,
means the symbol for the “union” in the set theory,
denotes the symbol for the “intersection”,
denotes the subset that contains all the labels observed by experiments for the k-th tested sample,
represents the subset that contains all the labels predicted for the k-th sample, and
(3)
In Equation (4), the first four metrics with an upper arrow
are called positive metrics, meaning that the larger the rate is the better the prediction quality will be; the 5th metrics with a down arrow 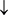 is called positive metrics, implying just the opposite meaning.
is called positive metrics, implying just the opposite meaning.
From Equation (2) we can see the following: 1) the “Aiming” defined by the 1st sub-equation is for checking the rate or percentage of the correctly predicted labels over the practically predicted labels; 2) the “Coverage” defined in the 2nd sub-equation is for checking the rate of the correctly predicted labels over the actual labels in the system concerned; 3) the “Accuracy” in the 3rd sub-equation is for checking the average ratio of correctly predicted labels over the total labels including correctly and incorrectly predicted labels as well as those real labels but are missed in the prediction; 4) the “Absolute true” in the 4th sub-equation is for checking the ratio of the perfectly or completely correct prediction events over the total prediction events; 5) the “Absolute false” in the 5th sub-equation is for checking the ratio of the completely wrong prediction over the total prediction events.
3.2. Comparison with the State-of-the-Art Predictor
Listed in Table 1 are the rates achieved by the current iATC_Deep-mISF predictor via the cross validations on the same experiment-confirmed dataset as
![]()
Table 1. Comparison with the state-of-the-art method in predicting iATC-mISFa.
aSee Equation (2) for the definition of the metrics. bSee [6], where the reported metrics rates were obtained by the jackknife test on the benchmark dataset of Supporting Information S1 that contains experiment-confirmed proteins only. cThe proposed predictor; to assure that the test was performed on exactly the same experimental data as reported in [6] for iATC-mISF.
used in [6]. For facilitating comparison, listed there are also the corresponding results obtained by the iATC-mISF predictor [6], the existing most powerful method for predicting the classes of anatomical therapeutic chemicals. As shown in Table 1, the newly proposed predictor iATC_Deep-mISF is remarkably superior to the existing state-of-the-art predictor iATC-mISF in all the five metrics. Particularly, it can be seen from the table that the absolute true rate achieved by the new predictor is over 67%, which is about 7% higher than iATC-mISF [6]. This is because it is extremely difficult to enhance the absolute true rate of a prediction method for a multi-label system as clearly elucidated in [6]. Actually, to avoid embarrassment, many investigators even chose not to mention the metrics of absolute true rate in dealing with multi-label systems (see, e.g., [10] [11]).
Meanwhile, as a byproduct, the present paper has also stimulated some very interesting or provoked papers (see, e.g., [12] - [17]).
3.3. Web Server and User Guide
As pointed out in [18], user-friendly and publicly accessible web-servers represent the future direction for developing practically more useful predictors. Actually, user-friendly web-servers will significantly enhance the impacts of theoretical work because they can attract the broad experimental scientists [19]. In view of this, the web-server of the current iATC_Deep-mISF predictor has also been established at http://www.jci-bioinfo.cn/iATC_Deep-mISF/, by which users can easily get their desired data without the need to go thru the mathematical details.
4. Conclusion
It is anticipated that the iATC_Deep-mISF predictor holds very high potential to become a useful high throughput tool in identifying the classes of anatomical therapeutic chemicals. Most important is that the predictor will become a very useful tool for fighting against the coronavirus to save mankind on this planet.
Acknowledgements
This work was supported by the grants from the National Natural Science Foundation of China (No. 31560316, 61261027, 61262038, 61202313 and 31260273), the Province National Natural Science Foundation of JiangXi (No. 20132BAB201053), the Jiangxi Provincial Foreign Scientific and Technological Cooperation Project (No.20120BDH80023), the Department of Education of JiangXi Province (GJJ160866).