Subject Areas: Analytical Chemistry, Organic Chemistry

1. Introduction
In the past few years, we have been involved in a program aimed at developing new efficient synthetic approaches for heteroaromatic compounds utilizing unexpensive starting materials [1] [2] . In continuation of our current interest in the syntheses of polyfunctionaly substituted heteroaromatic [3] - [6] , we used here the readily obtainable β-oxo anilide derivatives as starting materials. It is worthwhile to explore their potential utility for the synthesis of polyfunctionaly substituted pyridazinones, pyridine, pyrimidines and pyrazolotriazines.
2. Results and Discussion
It has been observed that coupling of 3-oxo-N-p-tolyl-butyramide 1 with diazotized aromatic amines in ethanol buffered with sodium acetate at 0˚C - 5˚C afforded the arylhydrazones 2a,b [7] . Arylhydrazones 2a,b were treated with N,N-dimethylformamide-dimethylacetal (DMF-DMA) in refluxing xylene to yield the pyridazinone derivatives 3a,b in good yield. The structure 3 was confirmed bases on spectral data, elemental analysis and their chemical transformation. 1H NMR spectrum for 3a revealed doublet at δ = 6.9 and 7.09 assigned for olefinic double bond (J = 2.0 Hz).
Compounds 3a,b were treated with hydrazine hydrate to yield a condensation product via water elimination as 4a,b. Compound 4a as example was established as the sole product based on elemental analysis and spectral data. Thus, IR spectrum showed absorption peaks at ν 3170, 3380 cm−1 for NH2 group and disappearance of peaks at ν 1684 cm−1 for CO group.
Reactions of 2a with hydroxylamine hydrochloride in ethanolic solution containing amount of sodium acetate afforded the condensation product 5 that analyzed correctly for C17H18N4O2. The structure of the latter product was identified as 2-(aryl-hydrazono)-3-hydroxyimino-N-p-tolyl-butyramide 5 on the basis of its IR and 1H NMR spectra. The 1H NMR spectrum displayed a two singlet signal at δ = 9.59 and 10.18 ppm assigned to two NH group. Trials to cyclized compound 5 to triazole 6 or indazole 7 under different condition failed (Scheme 1).
Similarly, the reaction of b-Oxo anilide 1 with hydroxylamine hydrochloride in aqueous ethanol in presence of sodium acetate give 3-hydroxyimino-N-p-tolyl-butyramide 8 not the pyrazolone 9 [8] . Compound 9 was ruled out by spectroscopic data. The IR spectrum of oxime 8 revealed ν cm−1 1652 for amidic CO; 3174 for NH; 3373 for OH.
The reaction of anilide 1 with aromatic aldehydes in the presence of ammonia has been reported to yield 2,6-dimethyl-1,4-dihydro-3,5-bis[(p-tolyl)carbamoyl]-4-(4-substituted phenyl)-pyridine 10a,b [9] . The structure of 10 was established bases on its correct elemental analysis and spectral data. Also, when compound 1 was reacted with a mixture of aromatic aldehydes and urea or thiourea, the tetrahydropyrimidines 11a,b was formed

Scheme 1. Synthesis of azo compounds and pyridazine derivatives.
[10] . The mass spectrum of 11b as example revealed a molecular ion peak at m/z = 371 corresponding to the molecular formula C19H18ClN3OS. Its 1H NMR spectrum showed a singlet signal at δ = 2.17 and 2.28 ppm assigned for the 2CH3 protons, a singlet signal (1H) at δ = 5.47 ppm assigned for pyrimidine-4H, a singlet at δ = 8.33, 9.22 and 9.97 ppm assigned for 3NH protons (Scheme 2).
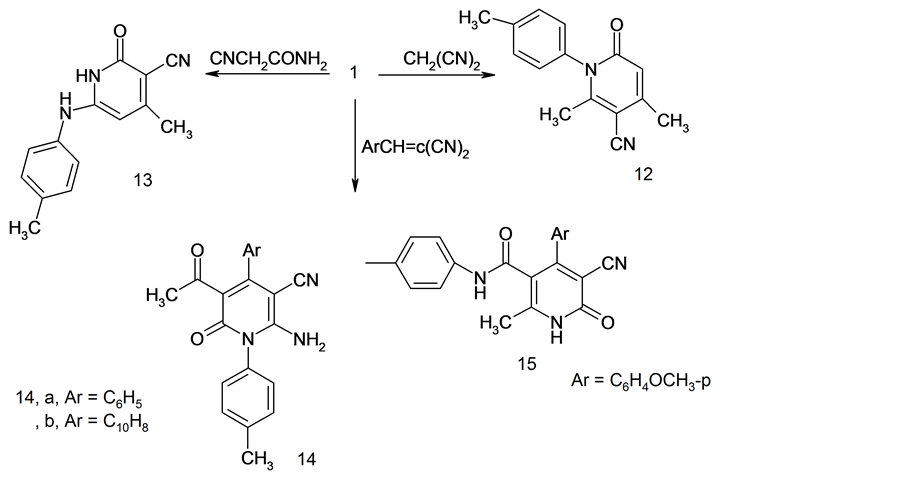
Reactions of compound 1 with active methylene reagents was also investigated. So, compound 1 was reacted with malononitrile in ethanolic piperidine to give the *yridine derivative 12. Structure 12 was supported by the appearance of NH2 absorption band at ν 3302, 3466 cm−1 in the IR spectrum. Moreover, its 1H NMR spectrum revealed a singlet signal at δ = 5.6 ppm assigned to CH-pyridine and 6.61 assigned to NH2 in addition to the other functional group protons.
Similarly, compound 1 was reacted with cyanoacetamide in the same experimental conditions to give 4-me- thyl-2-oxo-6-p-tolylamino-1,2-dihydro-pyridine-3-carbonitrile 13 in quantitative yield.
The reaction of 1 with arylidenemalononitrile depends on structure of substituent. Thus, reactions of 1 with benzylidenemalononitrile or naphthylidenemalononitrile afforded 14a,b while with p-anisidinemalononitrile afforded 15. Structure 14 was established bases on its spectroscopic data. Thus, the IR spectrum of 14b for example revealed absorption bands at ν 3420 and 3490 cm−1 assignable to NH2 group and ν 2245 cm−1 for CN group in addition to disappearance of band at ν 3260 cm−1 assignable to NH group. Assignment of structure 15 for the reaction product was based on its compatible spectroscopic data. Thus, its mass spectrum revealed a molecular ion peak at m/z = 373 (M+) corresponding to the molecular formula C22H19N3O3. Its IR spectrum showed absorption band at ν 3210 and 3300 cm−1 for (2NH), 2219 for (CN). Its 1H NMR spectrum revealed a two singlet signal at δ = 8.68, 10.06 ppm assigned for 2NH in addition to the other functional group protons (Scheme 3).
The active methylene group in compound 1 underwent an electrophilic substitution upon coupling with equimolar amounts of diazonium chloride of aminopyrazoles 16-18 afforded the expected pyrazolotriazines 19-21 respectively [11] [12] . These product compounds were established based on their elemental analysis and compatible spectroscopic data. Condensing 1 and aminopyrazoles 18b,c in refluxing isotropic water separator afforded the acyclic adduct 22a,b rather than the pyrazolopyrimidine 23a,b [13] . Assignment of structure 22 for the reaction product was based on its compatible spectroscopic data. Thus, its IR spectrum of 22a for example showed the presence of peak at ν 1665 cm−1 assigned for amidic carbonyl group (Scheme 4).
3. Experimental
All melting points are uncorrected. IR spectra (KBr) were recorded on a Ft IR 5300 spectrometer (ν cm−1). The 1H NMR spectra were recorded in DMSO-d6, CDCl3 at 200 - 400 MHz on a varian Gemini NMR spectrometer (δ, ppm) using TMS as an internal standard. Mass spectra were obtained on GC Ms-QP 1000 EX mass spectrometer at 70 ev. Elemental analyses were carried out by the Microanalytical Research Center, Faculty of Science at Cairo University and Microanalytical Research Center at Assiut University.
General procedure for preparation of compounds 2a,b.

Scheme 2. Synthesis of pyridines and pyrimidines.
Scheme 3. Synthesis of pyridine derivatives.

Scheme 4. Synthesis of pyrazolotriazine derivatives.
To a solution of compound 1 (0.01 mole) in dry xylene (10 ml), N,N-dimethylformamide-dimethylacetal (0.01 mole) was added. The reaction mixture was heated under reflux for 6 h. The solvent was removed by evaporation under reduced pressure and the remainder was left to cool. The solid product so formed was collected by filtration, washed with petroleum ether (b.p. 40˚C - 60˚C) and the crude product recrystallized from ethanol to give 2a,b.
4-Oxo-1-phenyl-3-(p-tolyl)carbamoyl-1,4-dihydro-pyridazine 2a.
This compound was obtained as brown crystals from ethanol; yield 72%; mp. 175˚C. IR (KBr) ν cm−1 1652 (CO); 1684 (CO); 2983 (CH-aliph.); 3410 (NH). 1H NMR (CDCl3) δ = 2.27 (s, 3H, CH3); 6.9 (d, 1H, CH); 7.09 (d, 1H, CH); 7.3-7.6 (m, 9H, Ar-H); 12.21 (s,
1
H
,
NH
). Found; C, 70.50; H, 4.70; N, 13.50; Calcd. For C18H15N3O2 (305.33): C, 70.81; H, 4.95; N, 13.76%.
4-Oxo-1-(p-chloro phenyl)-3-(p-tolyl)carbamoyl-1,4-dihydro-pyridzine 2b.
This compound was obtained as brownish crystals from ethanol; yield 72%; mp. 150˚C. IR (KBr) ν cm−1 1648 (CO); 1680 (CO); 2982 (CH-aliph.); 3350 (NH). 1H NMR (CDCl3) δ = 1.27 (s, 3H, CH3); 6.9 (d, 1H, CH); 7.1-7.68 (m, 8H, Ar-H); 7.7 (d, 1H, CH); 11.34 (s,
1
H
,
NH
). Found; C, 63.50; H, 4.00; N, 12.10; Calcd. For C18H14ClN3O2 (339.78): C, 63.63; H, 4.15; N, 12.37%.
General procedure for preparation of compounds 3a,b.
A mixture of 2a,b (0.01 mole) and hydrazine hydrate 0.5 ml in ethanol was refluxed for 6 hours. The product formed was collected and washed by cold ethanol to give 3a,b and recrystallized from proper solvent.
4-Hydrazono-1-phenyl-3-(p-tolyl)carbamoyl-1,4-dihydro-pyridazine 3a.
This compound was obtained as greenish crystals from ethanol; yield 62% mp. 230˚C. IR (KBr) ν cm−1 1648 (CO); 2975 (CH-aliph.); 3170, 3380 (NH2). 1H NMR (DMSO-d6) δ = 2.28 (s, 3H, CH3); 7.05 (d, 1H, CH); 7.92 (d, 1H, CH); 7.06-7.9 (m, 11H, Ar-H and NH2); 12.81 (s,
1
H
,
NH
). Found; C, 67.50; H, 5.20; N, 21.70; Calcd. For C18H17N5O (319.36): C, 67.70; H, 5.37; N, 21.93%.
1-(4-Chlorophenyl)-4-hydrazono-3-(p-tolyl)carbamoyl-1,4-dihydro-pyridazine 3b.
This compound was obtained as green crystals from ethanol; yield 62%; mp. 220˚C. IR (KBr) ν cm−1 1648 (CO); 2968 (CH-aliph.); 3380, 3456 (NH2). Found; C, 61.00; H, 4.40; N, 19.60; Calcd. For C18H16ClN5O (353.81): C, 61.11; H, 4.56; N, 19.79%.
General procedure for preparation of compounds 5 and 8.
A mixture of 2a,b or 1 (0.01 mole), hydroxylamine hydrochloride and sodium acetate in ethanol was refluxed for 6 hours. The solid product so formed was collected on heating, washed with cold ethanol and recrystallized from proper solvent to give 5 and 8.
3-Hydroxyimino-2-(phenyl hydrazono)-N-p-tolyl-butyramide 5.
This compound was obtained as white crystals from ethanol, yield 65% mp. 170˚C. IR (KBr) ν cm−1 1665 (CO); 2925 (CH-aliph.); 3280, 3299 (2NH). 1H NMR (DMSO-d6) δ = 1.53 (s, 3H, CH3); 2.25 (s, 3H, CH3); 2.34 (s, 1H, OH); 7.10 - 7.51 (m, 9H, Ar-H); 9.59 (s, 1H, NH); 10.18 (s, 1H, NH). Found; C, 65.50; H, 5.60; N, 18.10; Calcd. For C17H18N4O2 (310.36): C, 65.79; H, 5.85; N, 18.05%.
3-Hydroxyimino-N-p-tolyl-butyramide 8.
This compound was obtained as white crystals from dioxane, yield 60% mp. 360˚C. IR (KBr) ν cm−1 1652 (CO); 3174 (NH); 3373 (OH). Found; C, 64.10; H, 6.60; N, 13.50; Calcd. For C11H14N2O2 (206.25): C, 64.06; H, 6.60; N, 13.50%.
General procedure for preparation of compounds 10a,b.
A mixture of aldehyde (0.01 mole), compound 1 (0.01 mole) and 0.7 ml NH3 (25% w/v) were refluxed in 10 ml ethanol for 8-14 h. Cooled and then evaporated to dryness. The residue was crystallized from ethanol to give 10a,b.
4-(4-Chloro phenyl)-2,6-dimethyl-1,4-dihydro-3,5-bis[(p-tolyl)carba- moyl]-pyridine 10a.
This compound was obtained as white crystals, recrystallized from ethanol yield 70%; mp. 220˚C. IR (KBr) ν cm−1 1686 (CO); 3285 (NH). 1H NMR (CDCl3) δ = 2.27 (s, 12H, 4CH3); 7.0-7.41 (m, 12H, Ar-H); 8.0 (s, 2H, 2NH). Ms; m/z = 483 (M+). Found; C, 71.60; H, 5.30; N, 8.50; Calcd. For C29H26ClN3O2 (484.00): C, 71.97; H, 5.41; N, 8.68%.
2,6-Dimethyl-1,4-dihydro-4-(p-tolyl)-3,5-bis[(p-tolyl)carbamoyl]-pyridine 10b.
This compound was obtained as white crystals, recrystallized from ethanol yield 70%; mp. 195˚C. IR (KBr) ν cm−1 1655 (CO); 1699 (CO); 2925 (CH-aliph.); 3033 (CH-arom.); 3285 (NH). 1H NMR (DMSO-d6) δ = 2.23 (s, 6H, 2CH3); 2.33 (s, 6H, 2CH3); 2.49 (s, 3H, CH3); 7.05-7.51 (m, 12H, Ar-H); 8.45 (s,
1
H
,
NH
); 9.6 (s,
1
H
,
NH
). Found; C, 77.70; H, 6.30; N, 9.00; Calcd. For C30H29N3O2 (463.58): C, 77.73; H, 6.31; N, 9.06%.
General procedure for preparation of compounds 11a,b.
A mixture of compound 1 (0.01 mole), urea or thiourea (0.01 mole), p-Cl-benzaldehyde (0.01 mole), absolute ethanol (30 - 40 ml) and concentrated hydrochloric acid (8 - 10 drops) was stirred and slightly warmed on a steam bath till the mixture becomes a clear solution. It was allowed to stand overnight at ambient temperature. The product obtained was filtered off, dried and recrystallized from ethanol to give 11a,b.
4-(Chlorophenyl)-3,4-dihydro-6-methyl-5-(p-tolyl)carbamoyl-2(1H)-pyrimidinone 11a.
This compound was obtained as yellow crystals from ethanol; yield 80%; mp. 295˚C. IR (KBr) ν cm−1 1638 (CO); 1711 (CO); 3315, 3475 (2NH). 1H NMR (DMSO-d6) δ = 2.33 (s, 3H, CH3); 2.52 (s, 3H, CH3); 5.5 (s, 1H, 4(H)-pyrimidine); 7.0 - 7.75 (m, 9H, Ar-H and NH); 8.81 (s, 1H, NH); 9.91 (s, 1H, NH). Found; C, 64.00; H, 5.30; N, 11.70; Calcd. For C19H18ClN3O2 (355.83): C, 64.14; H, 5.10; N, 11.81%.
4-(Chlorophenyl)-3,4-dihydro-6-methyl-5-(p-tolyl)carbamoyl-2(1H)-pyrimidinethione 11b.
This compound was obtained as yellow crystals from ethanol; yield 78%; mp. 180˚C. IR (KBr) ν cm−1 1677 (CO); 3275, 3456 (2NH). 1H NMR (CDCl3) δ = 2.17 (s, 3H, CH3); 2.28 (s, 3H, CH3); 5.47 (s, 1H, 4(H)-pyrimi- dine); 7.0 - 7.42 (m, 8H, Ar-H); 8.33 (s, 1H, NH); 9.22 (s, 1H, NH); 9.97 (s, 1H, NH). Ms: m/z = 371 (M+). Found; C, 61.20; H, 4.70; N, 11.20; Calcd. For C19H18ClN3OS (371.89): C, 61.37; H, 4.88; N, 11.30%.
General procedure for preparation of compounds 12 and 13.
A mixture of 4 (0.01 mole), malononitrile (0.01 mole) and few drops of piperidine, was refluxed in ethanol for 4 h. The obtained solid on heating recrystallized from ethanol to give 12 and 13.
2-Amino-4-methyl-6-oxo-1-p-tolyl-1,6-dihydro-pyridine-3-carbonitrile 12.
This compound was obtained as white crystals from ethanol, yield 80% mp. 305˚C. IR (KBr) ν cm−1 1668 (CO); 2203 (CN); 3302, 3466 (NH2). 1H NMR (DMSO-d6) δ = 2.16 (s, 3H, CH3); 2.36 (s, 3H, CH3); 5.6 (s, 1H, CH-pyridine); 6.61 (s, 2H, NH2); 7.08 - 7.35 (m, 4H, Ar-H). Found; C, 70.00; H, 5.20; N, 17.30; Calcd. For C14H13N3O (239.28): C, 70.28; H, 5.48; N, 17.56%.
4-Methyl-2-oxo-6-p-tolylamino-1,2-dihydro-pyridine-3-carbonitrile 13.
This compound was obtained as white crystals from ethanol; yield 50%; mp. 170˚C. IR (KBr) ν cm−1 1658 (CO); 2217 (CN); 2951 (CH-aliph.); 3250, 3380 (2NH); Ms: m/z = 240 (M + 1). Found; C, 70.00; H, 5.20; N, 17.30; Calcd. For C14H13N3O (239.28): C, 70.28; H, 5.48; N, 17.56%.
General procedure for preparation of compounds 14a,b and 15.
To a solution of compound 1 (0.01 mole) in ethanol (40 ml) containing a catalytic amount of piperidine (0.5 ml), ylidenemalononitriles (0.01 mole) was added. The reaction mixture was heated under reflux for 6 h. The solid product formed on heating was collected by filtration to give 14a,b and 15.
5-Acetyl-2-amino-6-oxo-4-phenyl-1-p-tolyl-1,6-dihydro-pyridine 14a.
This compound was obtained as yellow crystals; recrystallized from ethanol yield 80%; mp. 202˚C. IR (KBr) ν cm−1 1648 (CO); 1696 (CO); 2190 (CN); 3210, 3280 (NH2). Found; C, 73.30; H, 5.00; N, 12.00; Calcd. For C21H17N3O2 (343.39): C, 73.45; H, 4.99; N, 12.24%.
5-Acetyl-2-amino-4-naphthalen-1-yl-6-oxo-1-p-tolyl-1,6-dihydro-pyridine 14b.
This compound was obtained as yellow crystals from ethanol; yield 80%; mp. 250˚C. IR (KBr) ν cm−1 1632 (CO); 1690 (CO); 2245 (CN); 3420, 3490 (NH2). 1H NMR (CDCl3) δ = 1.91 (s, 3H, CH3); 2.38 (s, 3H, COCH3); 4.31 (broad, 2H, NH2); 6.07 - 8.0 (m, 11H, Ar-H). Found; C, 76.10; H, 4.60; N, 10.50; Calcd. For C25H19N3O2 (393.44): C, 76.32; H, 4.87; N, 10.68%.
5-Cyano-4-(4-methoxyphenyl)-2-methyl-6-oxo-3-(p-tolyl)carbamoyl-1,6-dihydro-pyridine 15.
This compound was obtained as yellow crystals from dioxane; yield 72%; mp. 200˚C. IR (KBr) ν cm−1 1640 (CO); 1692 (CO); 2219 (CN); 3210, 3300 (2NH). 1H NMR (DMSO-d6) δ = 2.25 (s, 3H, CH3); 2.48 (s, 3H, CH3); 3.86 (s, 3H, OCH3); 6.9 - 7.42 (m, 8H, Ar-H); 8.68 (s, 1H, NH); 10.06 (s, 1H, NH). Ms: m/z = 373 (M+). Found; C, 70.50; H, 5.00; N, 11.00; Calcd. For C22H19N3O3 (373.42): C, 70.76; H, 5.13; N, 11.25%.
General procedure for preparation of compounds 19-21.
A solution of 1 (0.01 mole) in ethanol (100 ml) containing sodium acetate (2.0 g) was cooled to 0˚C, stirred and treated gradually with a cooled solution of aryldiazonium chloride (prepared from 0.01 mole of amine 16-18 and the appropriate quantities of HCl and NaNO2). The solid product formed on standing was collected and recrystallized from the appropriate solvent to give 19-21.
4-Methyl-7-phenyl-3-(p-tolyl)carbamoyl-pyrazolo[5,1-c][1,2,4]-triazine 19.
This compound was obtained as brown crystals from ethanol yield 60% mp. 220˚C. IR (KBr) ν cm−1 1670 (CO); 3256 (NH). 1H NMR (CDCl3) δ = 1.27 (s, 3H, CH3); 2.27 (s, 3H, CH3); 7.06 - 7.61 (m, 11H, Ar-H, CH- pyrazole and NH). Found; C, 69.80; H, 4.90; N, 20.10; Calcd. For C20H17N5O (343.37): C, 69.96; H, 4.99; N, 20.39%.
4-Methyl-7-oxo-3-(p-tolyl)carbamoyl-8-[4-(thiazol-2-ylsulfamoyl)- phenyl-azo]-6,7-dihydro-pyrazolo [5,1-c][1,2,4]triazine 20.
This compound was obtained as brownish crystals from ethanol; yield 70% mp. 220˚C. IR (KBr) ν cm−1 1640 (CO); 1695 (CO); 2907 (CH-aliph.); 3104 (CH-arom.); 3160 (NH). Found; C, 50.80; H, 3.20; N, 22.70; Calcd. For C23H19N9O4S2 (549.59): C, 50.27; H, 3.48; N, 22.94%.
7-Amino-4-methyl-3-(p-tolyl)carbamoyl-8-[4-(thiazol-2-ylsulfamoyl)-phenylazo]-pyrazolo[5,1-c][1,2,4]triazine 21.
This compound was obtained as brown crystals from dioxane; yield 60% mp. 300˚C. IR (KBr) ν cm−1 1655 (CO); 2925 (CH-aliph.); 3033 (CH-arom.); 3285, 3350 (NH2). 1H NMR (DMSO-d6) δ = 2.22 (s, 3H, CH3); 2.28 (s, 3H, CH3); 6.8 - 7.81 (m, 14H, Ar-H, CH=CH, 2NH and NH2). Found; C, 50.20; H, 3.50; N, 25.40; Calcd. For C23H20N10O3S2 (548.61): C, 50.36; H, 3.67; N, 25.53%.
General procedure for preparation of compounds 22a,b.
A mixture of 18b,c (0.01 mole), compound 1 (0.01 mole), ammonium acetate in acetic acid/benzene (10/30 ml) was heated under reflux (water separator) for 3h. The solvent was then evaporated under vacuo and the resulting solid products were filtered off and recrystallized from dioxane to give 23a,b.
3-[5-Amino-4-(4-sulfamoyl-phenylazo)-2H-pyrazol-3-ylimino]-N-p-tolyl-butyramide 22a.
This compound was obtained as brown crystals from dioxane; yield 55%; mp. 308˚C. IR (KBr) ν cm−1 1665 (CO); 3210, 3260 (NH2). 1H NMR (DMSO-d6) δ = 1.90 (s, 3H, CH3); 2.36 (s, 3H, CH3); 5.82 (s, 2H, CH2); 6.70 (s, 2H, NH2); 7.38 (s, 2H, NH2); 7.9-8.00 (m, 10H, Ar-H and 2NH). Found; C, 52.60; H, 4.60; N, 24.50; Calcd. For C20H22N8O3S (454.51): C, 52.85; H, 4.88; N, 24.65%.
3-(5-Amino-4-p-tolylazo-2H-pyrazol-3-ylimino)-N-p-tolyl-butyramide 22b.
This compound was obtained as brown from dioxane crystals; yield 59%; mp. 300˚C. IR (KBr) ν cm−1 1653 (CO); 3150 (NH); 3265, 3395 (NH2). Found; C, 64.60; H, 5.70; N, 25.10; Calcd. For C21H23N7O (389.46): C, 64.76; H, 5.95; N, 25.17%.
4. Conclusion
We are successful to synthesize a novel substituted pyridazinones, pyridine, pyrimidines and pyrazolotriazines.
NOTES

*Corresponding author.