Determination of Uranium Traces in Nuclear Reactor IEA-R1 Pool Water ()
1. Introduction
Uranium is an actinide element that its atomic number is 92. It occurs in natural water as traces, which rarely exceeds 30 µg/L. On average, the earth crust contains nearly about 4 mg/kg of uranium. Uranium, in the environment, occurs naturally as three radioactive isotopes: 238U (99.27%), 235U (0.72%) and 234U (0.005%). The isotope 235U is the only natural occurrence of fissile material [1] . As uranium is also important for its chemical and radiological properties, the modernity is dependent on this element to produce electrical power supply and nuclear medical radiopharmaceuticals through their fission products [2] . Nevertheless, uranium is toxic at sufficiently high levels to humans and the environment. As uranium has a great mobility throughout the Earth, it requires sensitive methods to trace quantities of this metal ion in the large spectrum of waters on Earth, along with that in plants, soils and rocks [3] .
Uranium has six oxidation states (0, +2, +3, +4, +5, and +6). The +4 state is relatively stable and is associated with hydroxides, phosphates, and fluorides. The +6 state is the most stable when present as octaoxide (U3O8 or yellow cake), but as hexafluoride it dissociates rapidly on contact with liquid water or water vapor in air [1] . Normally, the concentrations of uranium traces are in the VI oxidation state [4] .
In natural water, uranium occurs naturally normally dissolved as uranyl. While different national health authorities prescribe limits ranging up to 10 µg/L, the World Health Organization recommends a maximum concentration limit of 15 µg/L [U] for daily water consumption of 2 L per day; this limit leaves a considerable safety margin [1] .
The water feeding into the research nuclear reactors, such as IEA-R1 research reactor, comes from tap water, which receives a deep filtration and purification. One repeats this purification process continuously during reactor operation in order to keep the uranium trace level under control (<50 µg/L), besides other contaminants. Consequently, the water is safe for not provoking any uranium fission inside the reactor pool by nuclear reactions. The uranium-trace determination in the reactor water pool screens the purification process efficiency, since fuel elements may also contaminate the pool water with uranium residuals over fuel-element surface or more seriously by an unnoticeable fuel element fissure. The routine chemical method to evaluate uranium traces in pool water of IAE- R1 Reactor presently is by ICP-OES (Inductively Coupled Plasma Optical Emission Spectrometry), which has a limited detection level to the order of 30 μg/L.
Uranium is a very reactive element, readily combining with many elements to form a variety of complexes. The oxidized uranium compounds, as well, as the uranyl ion can combine easily with Cl−, Na3−, 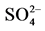 and
and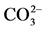 . In aerated aqueous solutions, at pH ≤ 3, uranyl ion is very stable. At higher pHs, uranyl ion tends to form stable complexes with phosphate and carbonate and precipitates. The information concerning uranium ion species, actually existing in the water supply, is critical for the selection of the treatment process and its successful operation [5] .
. In aerated aqueous solutions, at pH ≤ 3, uranyl ion is very stable. At higher pHs, uranyl ion tends to form stable complexes with phosphate and carbonate and precipitates. The information concerning uranium ion species, actually existing in the water supply, is critical for the selection of the treatment process and its successful operation [5] .
Various analytical methods are available to determine the presence, concentration, or quantity of uranium trace in a range of potable water (0.9 - 5 µg/L) until 200 µg/L [6] - [12] . Kuleff [13] , using methods of neutron activation, arrived to values of uranium content in primary coolant water of a PWR as 0.143 ± 0.017 µg/L [U].
Different complexing reagents are used in AdCSV technique (adsorptive cathodic stripping voltammetry) [14] , but with less discrimination of uranium, such as pyrocatechol, oxine, cupferron. Recent studies, summarized by Shrivastava, Sharma and Soni [15] , showed several complexing agents of uranium used in potentiometric measurements. Aluminon (3-[bis(3-carboxy-4-hydroxy- phenyl)-methylene]-6-oxo-1,4-cyclohezadiene-1-carboxylic acid triammonium salt) was used within the detection range of 2 - 33 µg/L, with discrimination of 0.2 µg/L. If this stripping technique is associated to square wave (SWAdSV), making use of propyl gallate, it was possible to measure in SPE microelectrode the range of 5 ng/L to 10 µg/L [3] . Sander, Wagner and Henze [4] used the AdCSV technique using chloranilic acid (CAA), which is chemically known as 2,5-dichlo- ro-3,6 dihydroxy-1,4-benzonquinone; they found the detection limit for this method as being around 24 ng/L. These authors concluded that AdCSV for uranium determination, using the electrolyte solution of 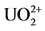 + CAA, acidified by H2SO4, is sensitive and selective method.
+ CAA, acidified by H2SO4, is sensitive and selective method.
Also, Dossi et al. [7] determined uranium traces in low-ionic-strength ground water. They used stripping voltammetry with chloranilic acid ligand. The authors stated that low-organic-content samples, no ultraviolet or oxidative pretreatments are necessary to be carried out. They measured a concentration range covering an interval between 80 to 145 µg/L.
Chloroanilic acid, according to Basavaiah and Charan [16] , has a formation of a charge-transfer complex between chloranilic acid as a π-acceptor and n-donor followed by the formation of radical anion according to the scheme presented in Figure 1.
Mustafa [8] identified the formation of the product trans- (n-Bu4N)2[UO2(CAA)2]∙3H2O, when adding [UO2(NO3)] in water to diluted chloranilic acid with diluted n-Bu4NOH. After infrared analysis, the author concludes that 2,5-dihydroxy-1,4-benzoquinone is an O,O donor. The chloranilate di-anion functions as an O,O ligand in trans-[UO2(CA)2]2−.
The present work aims at describe the use of AdCSV voltammetry process using CAA complex agent to evaluate IEA-R1 uranium trace level after a routine reactor shut down for maintenance and suggests a visual formation of UO2(CAA)2 monomer formation.
2. Experimental
To perform the evaluation of uranium trace in the pool water of IEA-R1 reactor,
![]()
Figure 1. Scheme for the formation of a charge-transfer complex [16] .
12 samples of 100 ml were collected from several points in the IEA-1 reactor pool. At the probing time, the reactor was shut down already for two days, preparing for a routine inspection. The samples were kept sealed in a fume hood at room temperature until the analyses were carried out.
The chosen voltammetric method was the adsorptive cathodic stripping voltammetry (AdCSV) using chloranilic acid (CAA) as the complex agent. The samples were acidified by H2SO4. These experiments in voltammetry were made in a Metrohm Voltammetric Analyzer VA797, which has advanced system capable to produce cyclic voltammetric stripping (CVS) measurements, using mercury micro-drops. Using differential pulse voltammetry, the setup parameters had the following data: initial purge time: 300 s; deposition potential: 0.025 V; deposition time: 20 s; equilibration time: 5 s; sweeping start potential: -0.010 V; end potential: -0.200 V; voltage step: 0.004 V; voltage step time: 0.100 s; sweep rate: 0.040 V/s; pulse amplitude: 0.050 V; pulse time: 0.040 s. Each voltammetric sample had 10 ml in volume. It was made an addition of 240 µL of 0.1 M H2SO4 to keep the acidity level within 2.5 < pH < 2.8. Addition of 120 µL of CAA (50 mM). Each experiment followed the sequence:
1- Five sweeping potential curves, in order to determine of cathodic current peaks and measuring each peak height in nA;
2- Two additions of 15 µL of standard solution with concentration of 10 µg/ml [U] followed by five sweeping curves each of them, producing two sets of five curves;
The evaluation of voltammetric response of chloranilic acid with uranium, as uranyl, was also studied by means of screen-printed carbon electrode (SPE) with a cyclic voltammetry (CV) study. The 80 µL sample containing CAA solution and uranium in presence of KCl was compounded by 40 µL of 10 mg/L uranium standard solution (acidified with H2SO4) + 20 µL of 50 mM CAA + 10 µL of 100 mM KCl. The acidity has approximately pH ~ 3. This drop sample was studied in a SPCE having silver as reference electrode and carbon as counter-electrode (silver acting as reference electrode in screen-printed electrode (SPE) in presence of KCl acquires the reference reduction potential as Ag/AgCl, between +0.200 to 0.220 VSHE, since the KCl concentration is not saturated). The 10-fold CV sweepings were made between −550 mV to +600 mV having a step potential a scan rate of 0.1 V/s. These measurements were made using Metrohm 910 PSTAT mini.
An evaluation of uranium level in homemade bidistilled water, which was used to prepare the uranium standard solution and CCA solution was also made in the Metrohm Voltammetric Analyzer VA797.
The effect of pH in experiments on hanging mercury drop, by means of differential pulse, was made using the exploratory system of Metrohm Voltammetric Analyzer VA797 using the same setup already described above.
3. Results and Discussion
The voltammetric analysis of homemade bidistilled water to certify the low level of uranium content was made in the VA equipment and revealed very low uranium content, as shown in Figure 2.
This result gave an average result of uranium content with the following regression line:
 (1)
(1)
This regression indicates that when current is zero, so no response of VA instrument, the amount of uranium concentration would be 0.055 µg/L. This level is an indication of remaining uranium content in water after bidistillation. This level represents a minor perturbation to final uranium content determination, as made for reactor pool water, which is in the range of one order more of magnitude.
The uranium determination by voltammetric techniques is not trivial, since many variables may arise from the experimental data. To qualify the broad behavior of UO2 + CAA, a ten-fold repetition cyclic voltammetry of CAA+U solution over a SPE was made, as shown in Figure 3.
As could be seen in the CV study of CAA + uranium solution, there is a fluctuation of the peaks of the curves around the cathodic potential of −0.09 V (CAA-uranyl complex precipitation) as CV repeats. In the anodic side, the reciprocal peak B, represents the dissolution of formed complex, as CAA2− and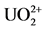 . This CV indicates a “quasi” reversible redox reaction between CAA and Uranium-IV (Uranyl), within the range of two involved electrons, since 0.59 V/electrons = 0.222, which theoretically, it should be 0.295. This redox CV did not pass all tests for a reversible system, as not reproducing the same Pa and Pc as the scan rate increased, but it is near. The involvement of approximately 2-elec- trons shows the effectiveness of a “quasi” redox reaction according to Mostafa
. This CV indicates a “quasi” reversible redox reaction between CAA and Uranium-IV (Uranyl), within the range of two involved electrons, since 0.59 V/electrons = 0.222, which theoretically, it should be 0.295. This redox CV did not pass all tests for a reversible system, as not reproducing the same Pa and Pc as the scan rate increased, but it is near. The involvement of approximately 2-elec- trons shows the effectiveness of a “quasi” redox reaction according to Mostafa
![]()
Figure 2. Analysis of homemade bidistilled water used to prepare the uranium.
![]()
Figure 3. Cyclic voltammetry curves of chloranilic acid in presence of uranium drop (around 50 µg/L), pH~3, acidified with H2SO4, over a SPE electrode (Metrohm E110), using carbon as working electrode and counter-electrode and silver as reference electrode. The numbers at the curves indicate the cycle repetition, where 1 is the first and 10 the last.
[8] explaining the chloranilate di-anion function. The first cycle in the CV, at the starting, a small bump (small peak B). It reveals that there was, before polarization, a small dissolution of the UO2-CAA complex, which had been formed in natural equilibrium. This demonstrates that the polarization towards peak A (cathodic direction), forming the reduced product of uranyl+CAA, is necessary to upheave the peak B, as displayed in the following sweepings. Just emphasizing, the CV at the anodic path oxidizes the complex back into its ions of 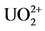 and CAA2− at peak B. Since there is same inertia to produce CAA2−, it is understood that same “training” is necessary to produce adequate amount of CAA2− to combine with the available uranyl.
and CAA2− at peak B. Since there is same inertia to produce CAA2−, it is understood that same “training” is necessary to produce adequate amount of CAA2− to combine with the available uranyl.
By mercury micro-drop voltammetry, there is a continuous change of drop and continuously renewing of cathode surface. Using one of the methods of voltammetry named hanging mercury drop electrode (HMDE), the formed complex is always remade at each sweeping, consuming the reactants. In SPE, there are some variations of peak heights from one repetition to the other, changing slightly the peak heights to positive direction. This is evident in Figure 3, where there is peak dislocation from the first to the tenth CV cycle, with lower cathodic-potential peak and higher anodic-potential peak. This is probably due to irregular diffusion of reactants near the SPE working electrode, since, in SPE simulation, the electrolyte is untouched during this experiment.
To analyze uranium content in AdCSV, the cathodic peak is used in the procedure, since it is the one adequate for cathodic potential position and responsiveness to uranium content in the sample.
It should be appointed that the peak of uranyl+CAA complex formation happen consistently between −0.05 and −0.11 V. The precise value for the potential of electrodeposition depends on pH, as shown in Figure 4.
In terms of acidity, the authors found that the most reactive electrolyte pH ranged between 2.4 until 2.8, where the voltammetric technique gave reproducible results and when the cathodic peak tends to grow at lower pH. There is, nevertheless, a compromise not to have a very low pH, since the determination curves start to penetrate into the anodic region, which is not reliable for the VA analyses, since the mercury may start oxidizing, giving erratic results.
Based on studies made by Boulet, Joubert, Cote, Bouvier-Capely, Cossonet and Adamo [17] and Mostafa [8] , a proposal for uranyl-CAA complex formation is indicated in Figure 5. This illustration indicates that the complex morphology may grow as a polymeric chain, becoming a stable substance covering the substrate during cathodic reduction of the reactants. This model suggests that the uranyl-CAA complex tends to be stable, not producing other derivative redox compounds with further polarization, either in cathodic or anodic polarization, as shown in Figure 3. Therefore, the complex does not decompose until the anodic polarization is carried out. This condition may impose a decrease the available amount of CAA and uranyl.
In the experimental tests, using IEA-R1 reactor water, the typical peaks manifested reproducible in the range of −70 to −110 mV. A typical sample plot for uranium trace determination in the voltammetry analyzer is shown in Figure 6. In these graph, there is a good set of curve replication in 3 phases of the process: sample analysis, after addition 1 and after addition 2, varying the sweeping potential towards the cathodic direction from −0.02 V to −0.20 V. This reproducibility is fundamental to have a reliable evaluation of uranium trace content with
![]()
Figure 4. Potentiometric curves showing the effect of acidity on CAA + UO2 electrolyte.
![]()
Figure 5. Schematic indication of the reaction of CAA and uranyl Complex.
![]()
Figure 6. One typical determination of U-content in IEA-R1 Reactor water and results of calibration of voltammetric determination (Sample 12).
a lower implicit error.
Table 1 lists the uranium content determinations for one of the typical reactor water sample (Sample 12). As could be seen in this table, it presents the mean results of sample and addition curves, as shown in the example given in Figure 6. A calibration line obtained gave the following equation for Sample 12:
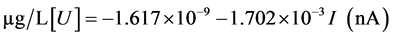 (2)
(2)
This typical statistical line has a mean standard deviation of 4.289 × 10−10 and a regression coefficient of R2 = 0.9997. In particular, based on this evaluation of Sample 12, the uranium trace content was 0.969 ± 0.034 µg/L [U]. Commenting
![]()
Table 1. Typical uranium content results of IEA-R1 reactor water.
VR = voltammetric sweepings in 3 levels (repetition is indicated in the second number); V = peak voltage; nA = current in nanoampere between the peak height and the base line; I.mean = mean of the nA; Std. Dev. = standard deviation of nA; I.Delta = difference the mean current between addition levels.
on the peaks of AdCSV results, there is a minor tendency to decrease the peak heights within each addition sequence, as could be seen in Table 1, confirming a stable formation of reduced CAA+UO2, which is lost with every mercury drop for each curve.
Table 2 summarizes the full experimental set, with all determinations, showing the data variation with 95% confidence standard error limits. As could be seen, the data dispersion varied from 0.798 to 1.070 µg/L [U] and the doubled standard deviation (95% of significance) varied randomly between 0.030 to 0.128 µg/L [U]. The mean value of all evaluations was 0.915 ± 0.190 µg/L [U].
A visual distribution of the uranium trace determinations in IEA-R1 pool- water samples, through the whole set, could be seen in Figure 7.
According to these results, the uranium-trace voltammetric results were consistent with normal indication in the range of regular tap water amount (0.8 - 2.0 µg/L) and presented no major concerns for having the possibility of uranium irradiation within the pool water for a major neutron capture. The maximum operating level for uranium level, in IPEN, is established as being < 50 µg/L. For more than 55 years, this maximum limit (50 µg/L) for uranium traces in IEA-R1 operation was not changed. Nevertheless, the mean evaluated trace level in this work (0.915 ± 0.190 µg/L [U]) showed that the pool water treatment was always adequate and operating under a substantial low uranium content level. It is possible to say that IEA-R1 Reactor has an adequate and reliable pool-water purifying system, promoting no major risk to reactor operation in terms of free
![]()
Figure 7. Dispersion of uranium traces content of 12 poll-water samples of IEA-R1 reactor.
![]()
Table 2. Results of voltammetric analyses of uranium traces in pool water of IEA-R1 reactor.
uranium in the pool water. As the purifying system never changed, probably the IEA-R1 reactor always had a good quality for its water.
4. Conclusions
The present study scrutinized uranium traces of IEA-R1 pool-water by statistical sampling and analyses. The employed methodology to determine uranium was adsorptive cathodic stripping voltammetry (AdCSV) using chloranilic acid complex (CAA). The electrochemical deposition of uranyl-CAA complex over the mercury drop was found to be based on chain formation between uranyl cation and chloranilic ring by means of oxygen-bridge, confirmed by cyclic voltammetry that chloranilate di-anion functions as an O,O ligand in trans-[UO2(CA)2]2−. A visual model for this combination was suggested in this work. The ACSV method using CAA to determine uranium content, confirmed the literature as a reliable determination. This method revealed reproducible allowing to verify that IEA-R1 pool-water is working in the uranium-trace level of 0.915 ± 0.190 µg/L. This level is far less than the functional established level < 50 µg/L [U], which has been considered as a technical limit for not promoting nuclear hazard under reactor operation.
Acknowledgements
Thanks are due to Fundação de Amparo à Pesquisa do Estado de São Paulo― Project FAPESP 2013/08514-3 for supporting financially this scientific work.