Hydrogeochemical Relationships between Spring and Subsurface Waters in the Dindefello Area of South Eastern Senegal ()
1. Introduction
This study aimed at evaluating the hydrogeochemical relationships between spring and subsurface groundwater occurring in the Dindefello area, a highlands region located on the Senegal-Guinea border. The study idea stems from the observations (made by the authors during a routine geology field trip mapping for students) that spring discharges avalanche down onto the valley floor, drains and infiltrate the ground, thereby recharging the ground- water and forming a major source of groundwater on an annual basis. The question we addressed was: which hydrogeochemical processes might influence the hydrochemical evolution of the groundwater as it was being recharged by the infiltrated spring water. The interpretative approach was to determine the major ions composition of both spring and groundwater, and analyze them for any correlation and mineral-water relationships.
2. Characteristics of the Study Area
The study area is a crystalline environment located on the Senegal-Guinea border (Figure 1). Administratively, it is part of the Bandafassi prefecture (Kedougou Region), a highlands area with a series of isolated valleys and hills with average heights of about 200 m. Highest crests are around 440 m above the surrounding plain areas. Figure 1 shows a generalized topographic setting of the area, with 40 m contour interval.
Perennial topography-driven springs occur in the study area, which can be classified according to their location, into: Type I (those that are located on altitude and are cascading down the side walls of hills as waterfalls onto the valley floors, e.g.: Dindefello, Dande and Nandoumari cascades); and Type II (those that are discharging at the foothills, and may flow toward the drainage network or build up swampy environments (Pr 1, 2, 3 and 4). Moreover, there are water-supply pondlets dug by villagers in river beds which are classified as Type III.
Highest spring flow rates occur during the rainy season and decreases subsequently in the dry season. Water supplies for human subsistence and cattle ranching are usually obtained from shallow hand-dug wells bored in the upper aquifer unit (i.e. weathered overburden). The hand-dug well yield is typically small (<0.2 m3/h). During the dry season, however, the depth to groundwater level usually increases rapidly leading to water-supply shortage problems. To meet the water need, the inhabitants rely on the water from springs and pondlets to complement the drying shallow-well water.
Geologically, the study area is underlain by basement rock of the so-called “Ségou group” (Upper Proterozoic to Lower Paleozoic age). Descriptions of the geology can be found in [1] -[4] . The rock outcrops build up a topographic mountainous setting consisting, in order upwards, of the Kafori Formation, the Pelel Formation and the Dindefello Formation. The basal Kafori formation, which is used as a stratigraphic marker, is chiefly basement pebble-conglomerate that lies unconformably over parental basement rocks. The Pélel Formation consists
![]()
Figure 1. Location map showing the study area within southeastern Senegal and topographic setting.
of the alternation of feldspathic sandstones (bed thicknesses of 10 - 60 cm) and calcareous pelites (mudstones/ siltstones) interbeds (1 - 10 cm thick). In outcrop, the base of the overlying Dindefello Formation mainly consists of decimetre-scale to several decimetre-thick bedded fine- to medium-grained feldspathic sandstone bars with minor inter-bedded pelite layers. The uppermost part of this formation evolves to thinner (centimetre to decimetre thick) bedded argillocalcareous sandstone bars with thin clay interbeds. The sedimentation typically forms a fining upward facies succession that passes from rudites through arenites and lutites to siltites. All these formations have been intruded by volcanic rocks (Carboniferous to Jurassic) that added to faulting and fracturing the host rocks.
The soils support a degraded savannah vegetation type. The climate is humid, with alternating dry seasons (from November to April) and wet seasons (from May to October). Average annual rainfall is about 1200 mm and precipitation typically exceeds potential evapotranspiration during the rainy season. Regional water budget estimates, based on empirical methods [5] , suggest a net rainfall of about 19% - 21% of the annual precipitation.
Direct infiltration of rainwater via the subsurface soils is not likely to spiel a predominant role in the study area due to the clayey character of the overburden material. Groundwater is recharged locally and essentially by rain and springwater runoff via the alluvium of river channels and their tributaries. The groundwater aquifer in this area comprises an upper water-bearing zone (shallow aquifer) of poor permeability material which typified the soils developed from the underlying bedrock. It is an admixture horizon of sand, gravel, silt and clay, with the water table mostly encountered at depth of 10 - 30 m below ground surface. Hydrogeologically, this upper unit is hydraulically connected to the underlying zone of bedrock disintegration and leaching, which forms a more productive zone reservoir.
3. Methods
This study is based on hydrochemical analyses carried out on water samples taken in May 2008 during a 14-day field mapping training course for students of our institute. The sampling distribution is presented in Figure 2. A total of fifteen water samples were collected out of which 8 were from springs’ discharges, 5 from nearby village groundwater supply hand-dug wells drilled in the weathered bedrock, and 2 from water supply pondlets drilled in streambeds. Sampling depths ranged from 0 m for spring water samples, from 1 - 2 m for pondlets groundwater samples, and from 10 - 20 m for groundwater sampled from hand-dug wells. During the sampling, the samples were first collected in 1 L plastic bottles, almost to a spilling level (thus avoiding bubbles), and was then transferred into 250-mL propylene bottles for storage, after filtering through Millipore 0.45 mm filters. For each sample the replicate for cation determination was preserved with Concentrated HCl 0.1-N to pH of £2, before storage (following cooling in an ice bath). Field physico-chemical measurements were made for pH, temperature and electric conductivity (EC).
![]()
Figure 2. Sampling locations. Note: Refer to Figure 1 for the location of Pr 13 (Tiangué Village).
The samples were analyzed by the conventional technique of ion chromatography (IC) for Ca2+, Mg2+, Na+, K+, Sr2+, Fe2+, Al3+, B3+ ions as cations, 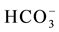 , Cl−,
, Cl−, 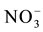 ,
,  , F−,
, F−, 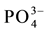 ions as anions, and for nonionic solute silica (SiO2) at the laboratory of the Institute of Geology and Paleontology of the University of Muenster (FRG). The analyses for groundwater samples were performed in almost all cases with better than ±10% precision, the acceptable limit indicated by [6] , except in one case (sample Pr 5). For springwater samples which exhibited near rainwater composition, the charge balance errors frequently exceeded 20% which is, according to [6] , a familiar problem when analyzing very low mineralized waters, such as rain water. However, these analytical errors were not very influential on the interpretation, as will be seen below.
ions as anions, and for nonionic solute silica (SiO2) at the laboratory of the Institute of Geology and Paleontology of the University of Muenster (FRG). The analyses for groundwater samples were performed in almost all cases with better than ±10% precision, the acceptable limit indicated by [6] , except in one case (sample Pr 5). For springwater samples which exhibited near rainwater composition, the charge balance errors frequently exceeded 20% which is, according to [6] , a familiar problem when analyzing very low mineralized waters, such as rain water. However, these analytical errors were not very influential on the interpretation, as will be seen below.
4. Results and Discussion
4.1. Origin of Major Ion Chemistry
The analytical results are summarized in Table 1. The relative participation of the main solute constituents to total mineralization as indicated by total dissolved solids (TDS) ranges between 23 and 567 mg/l. Conductivity values range from 24 to 780 mS/cm. pH values are between 4.65 and 6.85 indicating a slightly acidic water for the samples. A striking feature of springwater samples is their very low mineralization grade (TDS £ 63 mg/l) which strongly resembles that of rainwater (Table 1 provides a summary comparison of average chemical composition of rain and springwater). Low mineralization of springwater could be due to a rapid flow of springwater through the fractures and fault zones; this would mean that springwater is, on the one hand, either interflow or single-season water; or, on the other hand, it is due to its short residence time, being the recently infiltrated rain- water. However, the rainy season had not set in at the sampling time and springwater probably flows so fast that there is not enough contact time for an effective reaction with the host rocks.
All concentrations are in mg/l. *TDS (mg/L) = Total dissolved solids content. **Mean of monthly compositions of rainfalls reported by [7] in the region; viz.: chemical composition of rainfalls in july, august and September 1981 at the climatic stations of Tambacounda and composition of rainfalls in july and augsust 1981 at Kédougou station. ***Mean composition of spring water by ignoring the two cases of anomalous higher mineralized spring waters. Abbreviation: n. a: not analyzed.
The contrast between springwaters of low concentration and the more concentrated spring or pondlet waters suggests an increase in TDS due to the influence of marshy conditions prevailing at some sampling locations. Springwater samples that possess hydrochemical fingerprint that deviated from this pattern are attributed to those originated from poorly drained swamps that are floored with mud and plant debris which promote chemical reactions. Under these conditions, water may be acidic from CO2 production from organic matter decay.
Moreover, the TDS ranges for groundwater (usually below 600 mg/l) strongly suggest a recent history of origin, to conceivably represent the very early initial stage (Phase I) of groundwater mineralization process. The term Phase I is used here to indicate that a later Phase II is possible during confinement (i.e. with aging), during which ionic exchange reactions and ferric sulfides reduction may occur [5] .
The important charge balance components (i.e. the constituents that amount to more than 5% of the TDS) are , SiO2, Ca2+, Mg2+ and Na+. They average almost 80% - 99% of the TDS and are seldom lower than this, except in the case of water samples with elevated nitrate (up to 21%) and/or chloride contents (up to 5%), both related to contamination. These five elements are distributed in range 17% - 45% of the TDS for
, SiO2, Ca2+, Mg2+ and Na+. They average almost 80% - 99% of the TDS and are seldom lower than this, except in the case of water samples with elevated nitrate (up to 21%) and/or chloride contents (up to 5%), both related to contamination. These five elements are distributed in range 17% - 45% of the TDS for , 8% - 52% for SiO2, 7% - 40% for Ca2+, 2% - 18% for Mg2+ and 2% - 14% for Na+. It should be noted that K+ contributes, at best, 3% of the total mineralization compared to Na+ and often scores less than 2%. The higher mineral contents observed in groundwater samples may be regarded as the consequence of a greater magnitude of water- rock interactions along groundwater flow paths in the aquifer where the major ions’ mineral phase-contributors undergo dissolution. So, judging from the observed solute constituents association, and our field observations, we could probably suggest that groundwater in the shallow subsurface derives its mineralization essentially through the processes of carbonates and silicate minerals breakdown. The following discussion describes this possibility through the results of analyzed data for diagram plotting, correlation and mineral-water relationships.
, 8% - 52% for SiO2, 7% - 40% for Ca2+, 2% - 18% for Mg2+ and 2% - 14% for Na+. It should be noted that K+ contributes, at best, 3% of the total mineralization compared to Na+ and often scores less than 2%. The higher mineral contents observed in groundwater samples may be regarded as the consequence of a greater magnitude of water- rock interactions along groundwater flow paths in the aquifer where the major ions’ mineral phase-contributors undergo dissolution. So, judging from the observed solute constituents association, and our field observations, we could probably suggest that groundwater in the shallow subsurface derives its mineralization essentially through the processes of carbonates and silicate minerals breakdown. The following discussion describes this possibility through the results of analyzed data for diagram plotting, correlation and mineral-water relationships.
4.2. Evidence of Hydrochemical Processes
4.2.1. Hydrochemical Facies
Figure 3 shows two diagrams for presenting the major element characteristics of the sampled waters. The Schoeller diagram (Figure 3(a)) provides a quick view on trends (namely, contrast and similarity) in the concentration of dissolved ions, while the Piper-diagram (Figure 3(b)) enables the grouping of the data into water types, whereby the circle radius is a scale for TDS. Most samples plot in the soft water domain as represented by calcium magnesium bicarbonate (Ca-Mg-HCO3); this is typical of shallow groundwater or recharge water into a geological environment containing carbonates minerals. However, two samples (Pr 11 and Pr 13) are of a sodium bicarbonate (Na-HCO3)-dominated type, which suggests an alkali-ions water type, possibly resulting from contribution from sources other than common salts. It is interesting to note that some minor nitrate and chloride impairment (possibly from human impact) causes springwater samples to vaguely shift along the [SO4 + Cl + NO3]-axis of the lozenge. However, any impairment of the water quality which may give cause for health concern is noticed.
4.2.2. Linear Regression Study
Table 2 highlights the relationships between variables. The elements that correlate significantly (95.0% confidence level of significance) are indicated in bold character. As can be seen, all relevant charge balance constituents correlate with the TDS. Moreover, most of them inter-correlate, suggesting the same mechanism of origin. Apparently, the simultaneity of their source mechanisms ensures that the solute constituents making up the bulk of groundwater mineralization are largely “dependent variables”, all correlating significantly.
In essence, 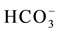 correlates well with all major constituents but, in apparent order of decreasing strength, better with Sr+ (p-value = 0.0003), Ca2+ (p-value = 0.0003), Mg2+ (p-value = 0.0006), F− (p-value = 0.0032). It seems likely that such a grouping arises possibly because these elements tend to accumulate from carbonates dissolution. Also, the dependence between
correlates well with all major constituents but, in apparent order of decreasing strength, better with Sr+ (p-value = 0.0003), Ca2+ (p-value = 0.0003), Mg2+ (p-value = 0.0006), F− (p-value = 0.0032). It seems likely that such a grouping arises possibly because these elements tend to accumulate from carbonates dissolution. Also, the dependence between  and both Ca2+ and Mg2+ might indicate ionic contribution from sulfate dissolution. The fact that dissolved SiO2 correlates significantly with this group of elements provides an additional indication that related factors control their occurrence. It is worth noting, however, that the strongest correlations to emerge for SiO2 are with K+ (p-value = 0.0041), Na+ (p-value = 0.0125), F− (p-value = 0.0121), Ca2+ (p-value = 0.0194) and Al (p-value = 0.026); an indication that most of these elements may have been leached from silicate mineral breakdown. This view is supported by the positive correlations that come into view between the pH values and almost all major water constituents (see Table 2).
and both Ca2+ and Mg2+ might indicate ionic contribution from sulfate dissolution. The fact that dissolved SiO2 correlates significantly with this group of elements provides an additional indication that related factors control their occurrence. It is worth noting, however, that the strongest correlations to emerge for SiO2 are with K+ (p-value = 0.0041), Na+ (p-value = 0.0125), F− (p-value = 0.0121), Ca2+ (p-value = 0.0194) and Al (p-value = 0.026); an indication that most of these elements may have been leached from silicate mineral breakdown. This view is supported by the positive correlations that come into view between the pH values and almost all major water constituents (see Table 2).
![]() (a) (b)
(a) (b)
Figure 3. Schoeller (a) and Piper diagram (b) of the samples. Key: In Figure 3(b), circles represent groundwater samples, and triangles are spring water samples.
![]()
Table 2. Matrix diagram displaying the Spearman rank correlations coefficients between variables.
Note: Correlations that are consistent at the 95.0% confidence level are indicated in bold character.
We speculate from this inference that the pH-driven process of minerals breakdown operating in the vadose zone under the leaching action of H+ is the main control of groundwater mineralization in the subsurface environment of the study area. In this respect, there are significant inverse (i.e. negative) correlations between Al3+ and pH on the one hand, and between Al3+ and both Na+ and SiO2 on the other hand (i.e. Al3+ correlates also with K+ negatively, but not significantly). This is in contrast with the positive correlations between pH and Na+, K+ and SiO2, a situation that can be interpreted as probably representing how feldspars (mostly Na-feldspars) dissolve upon consumption of H+ to produce dissolved ions or ion complexes and other secondary minerals products (like kaolinite, silicic acid and silica). The way albite dissolves through hydrolysis can be illustrated as follows:
Step 1: 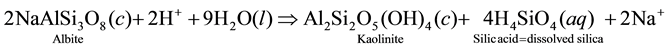
Step 2: 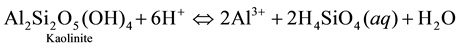
Step 3: ![]()
Also, it cannot be excluded that K-feldspar (KAlSi3O8) also dissolves. However, most of our attention is focused on Na+, which is by far the most abundant alkali ion in solution. Na-feldspar is, according to [8] , much more reactive than K-feldspar. In addition, it worth noting that K+ ions that could be released from K-feldspar mineral breakdown may be subjected to preferential K-chelation by organic compounds in the soil humus, thus sequestering this ion in a form that could be available for plant growth. Such a selective removal of K+ relative to Na+ would result in higher Na+ concentration in groundwater. Fluoride (F−) and strontium (Sr2+) correlate together and with all alkali metal ions suggesting that they owe their origin to parallel processes. Finally, the negative correlation between bromide (Br−) and the dominant solute constituents suggests a tendency of this element to deplete as mineralization progresses, i.e. dissolved Br− ion may afterward be adsorbed by the clay-membrane effects.
Finally, we note that the concentrations of Al3+ are negligible. Measurable amounts of Al3+ ions were found only below pH 6 in silica (SiO2) depleted water samples, thus substantiating the tendency of Al3+ to go into solution as acidity augments (viz., according to step 2 of the albite decomposition illustrated above). On the other hand, Al3+ tends to precipitate at higher pHs where silica and other by-products are more soluble ( [9] , [10] ). This raises the possibility that it may be kaolinitic by-products that incorporate Al3+ removed from feldspars dissolution, as suggested by the results of plotting the water composition in the stability diagram for kaolinite (see Figure 4).
![]()
Figure 4. Korinjski diagram of the water samples.
The proposed mechanism, as described in most groundwater textbooks (e.g., [11] -[17] ), considers that carbon dioxide present in natural waters (H2CO3) is the main source of H+, and so exerts the dominant pH control in subsurface groundwater. H2CO3 can release H+ ions through the reaction steps: ![]() and
and ![]() . (Here, the role of others weak acid groups in affecting the pH is neglected, notably that of organic acids.)
. (Here, the role of others weak acid groups in affecting the pH is neglected, notably that of organic acids.)
The postulated mechanism assumes that H+ ions thus released during the course of groundwater flow react with the aquifer sediments to produce the observed groundwater chemistry. In this connection, we now examine the saturation state for reactions that may control the water chemistry in the aquifers (see Table 3).
4.2.3. Equilibrium Solubility Study
As a check for the proposed model reactions, mineral speciation were calculated for the water samples using “Diagrammes” (version 5.1), a freely down-loadable software program of the University of Avignon (www.lha.univ-avignon.fr/). The calculations provided saturation indices (SIs) for possible minerals that may be reacting in the aquifers to give the observed water chemistry. Agreement is shown by the model results (Table 3) that, in contrast with springwater, groundwater is nearing saturation with respect to calcite, dolomite, aqueous silica and dissolved CO2, thus indicating their potential control on groundwater chemistry. We use here the term “saturation” in the sense of Langmuir [16] ; i.e., −0.1 £ SI £ + 0.1. However, nearly all SI-vales are negative, indicating that almost all these minerals are apparently unstable within the study area and may be actively undergoing dissolution. Calcite and gypsum exhibit more positive SI-values than their respective counterparts (namely, aragonite and anhydrite), thus indicating that they are more likely solute contributors to the groundwater chemistry. Quite evidently, the controlling effect of dissolved CO2 on the solubility is also apparent from the table.
It seems probable, however, from the Korinjski stability diagram (Figure 4) that dissolution of albite in the process of weathering-forming kaolinite exerts control over the concentrations of Na+ and silica (expressed in terms of H4SiO4). A general trend of concentrations increase in these constituents is apparent. Groundwater visibly plots close to the albite-boundary, which means that it is nearer to equilibrium with respect to this mineral as mineralization proceeds.
![]()
Table 3. Results of the aqueous speciation calculations for the samples.
Note: Numerical values are the saturation indices of minerals which may influence the water chemistry in the aquifers.
4.2.4. Correlations
The sample plots of Figure 5 provide a useful check on how well the geochemical model evolution conforms to the data. The general trends of the curves are similar, indicating systematic increases in bicarbonate, calcium and magnesium as groundwater is approaching saturation with respect to both calcite and dolomite. In this figure, the data points exhibit an orderly distribution, with springwater samples plotting on the lower part of the profile and groundwater samples on the upper part. This data scattering generally coincided with a spatial disposition of the samples, consistent with the spatial variability in the terraine lithlogies. Highest concentrations (TDS > 490 mg/L) occur downgradient the western part of the study area (Pelel-Pelou sectors) where the groundwaters are calcium-carbonate dominated. Moreover, the highest Sr2+ and F− contents are found there, also indicating a similar geochemical origin.
As may be seen from Figure 6, increasing Sr2+ and F− concentrations occur as groundwater is nearing saturation with calcite, probably due to the limey nature of the substratum in this zone. The highest Sr2+ and F− contents occur down-slope the south and western flanks of the Nandoumari Heights, in the Tanague-Pellou sector plain (samples Pr 5, 9 and 15) where carbonates and dolomites formations outcrop. Down-slope to the northeast, where weathering has developed on granitic birimian basement (Mamadou Boundou and Dindefello plain areas), the aquifer sediments appear to be less calcareous; hence the groundwater there are lower in dissolved solids content (less than 250 mg/L), and mostly sodium-bicarbonate dominated.
![]()
Figure 5. Comparison of calcite and dolomite saturation indices as a function of dissolved bicarbonate, calcium and magnesium contents.
The most probable explanation for the correlation shown in Figure 6 is that strontium and fluoride accumulate in the carbonates. The most probable origin for F− is from dissolution of fluorapatite (Ca5(PO4) 3F ) that accumulated from organic activity in the geologic environment during sedimentation. This explanation is reasonable because phosphate typically is a minor constituent of most limestones and limey mudstones.
The possibility of gypsum dissolution contributing sulfate and calcium ions is illustrated in Figure 7. However, gypsum solubility is seemingly very low, meaning a very small contribution to the observed major ion chemistry. Figure 8 provide a good appreciation for explaining why the occurrence of alkali-ions is not associated with chloride (i.e., alkali-ions from sources other than salts) but could be due to contributions from both cationic exchange (as may be seen from Figure 8(a)) and feldspars hydrolysis (Figure 8(b)). The fitted regression curves likely suggest that these two processes could occur simultaneously during mineralization.
![]()
![]()
Figure 6. Calcite saturation index as a function of strontium and fluoride concentrations.
![]()
![]()
Figure 7. Plot of gypsum saturation as a function of dissolved sulfate and calcium.
4.3. Model of Geochemical Evolution
The characteristically very low degree of groundwater mineralization clearly suggested a recent history of origin; that is, recharge had occurred from most recent rainfall events. Accordingly, one may consider the proposed mechanism of groundwater mineralization to conceivably represent the early stage evolution of groundwater chemistry through the pH-driven process of minerals breakdown under the leaching action of H+ ions.
A model has been proposed to explain the hydrochemical history of shallow groundwater as follows. Soils in the recharge area supply CO2 to seeping water, thus supplementing the amount of aqueous CO2 (H2CO3) already entrapped from the atmosphere. Then H+ ions, produced from dissociation of aqueous CO2 (![]() ) during the course of infiltration and underground flow, react with the aforesaid rock minerals, resulting in the observed major ion groundwater composition.
) during the course of infiltration and underground flow, react with the aforesaid rock minerals, resulting in the observed major ion groundwater composition.
The controlling effects of pH and dissolved CO2 on the solubility of minerals can be illustrated as follows (thereby limiting the discussion to a qualitative reasoning based on the example of carbonate of calcium, dolomite and albite). Dissolved CO2 has long been recognised as a key variable in the major ion loading of carbonate groundwaters ([18] -[21] ). Therefore, when a weak acid like H2CO3 dissolves for example calcium carbonate or dolomite, the overall process may be summarized by the following reactions:
![]()
![]()
The decomposition of albite can be represented by the reaction with water containing H+, as ([6] , [11] ):
![]()
The kaolinite produced by this reaction may dissolve reversibly under some conditions ( [22] ), following the reaction:
![]()
The dissolved silica may be precipitated in turn if high concentrations are reached:
![]()
Thus, any process that tends to increase the amount of CO2 available in groundwater (or decrease its pH) makes more minerals dissolve.
5. Summary and Conclusions
This study was carried out to investigate the hydrogeochemical processes prevailing in the Dindefello Plain area, a poorly documented hydrological system. Fifteen water samples were taken from the area for classical hydrochemical analysis. The results of integrated field observations, together with the plotting of key diagrams, correlation analysis and aqueous speciation, provide a basis for identifying the processes of groundwater mineralization. The following hydrogeochemically significant conclusions concerning the interaction between spring-, groundwater and rock within the aquifers are drawn:
• Springwaters contributing to groundwater recharge in the study area reflected a near-rainwater chemical composition which is an indication of recent recharge from the precipitation in this zone;
• Shallow groundwater typically showed bicarbonate, silica, calcium, magnesium and sodium to be about 80% - 99% of the total mineralization. Spatial variations in the groundwater chemistry are discernible suggesting that the type of bedrock aquifer (in terms of its chemical and mineralogical characteristics) has an effect on the water chemistry. In general, groundwater from the western part of the plain area tend to have much higher total dissolved solid contents and concentrations of fluoride and strontium than do samples from the eastern part; and
• The major hydrochemical mechanisms identified in this study, and which determine the chemical change in the aquifers within the study areas, are: 1) carbonate dissolution; and 2) feldspar dissolution, under the leach- ing action of H+ from dissolved carbon dioxide. Evidence from Mechanism 1 is the dominance of calcium- bicarbonate water type. Mechanism 2, which is relatively lower than Mechanism 1, tends to increase the Na and aqueous silica concentrations in groundwater. Calcium-for-sodium cationic exchange (i.e. Mechanism 3) that would act from mineral weathering is possible, as indicated by the occurrence of sodium-bicarbonate water type.
Further studies are to be carried out in this area using isotope hydrochemical approach to better understand the complex interrelations between spring and ground waters of the study area.