Process for the Preparation of Chromones, Isoflavones and Homoisoflavones Using Vilsmeier Reagent Generated from Phthaloyl Dichloride and DMF ()
1. Introduction
In recent years, scientific interest towards chromones (2), isoflavones (9) and homoisoflavones (10) has increased. It is due to the limited distribution of these compounds in the plant kingdom and the possible health effect these compounds exhibit. The development of new methodologies for the synthesis of these compounds is important. It is known that certain natural and synthetic chromone derivatives possess important biological activities such as antitumor [1] , antihepatotonic, antioxidant [2] , anti-inflammatory [3] , antispasmolytic, estrogenic [4] and antibacterial activities [5] . Isoflavones are a privileged class of natural products which are produced by plants mainly in the species of Leguminosae family to protect themselves from environmental stress and are present in dietary components such as fruits, cabbage, soybeans, grains, hops and redwines. Isoflavones possess many biological activities such as estrogenic [6] , anticancer [7] , antibacterial [8] , antimicrobial [9] , antiulcer [10] and protein tyrosine kinase inhibitor [11] . Search for new methodologies for the synthesis of isoflavones continues to be of great interest for organic chemists. The two most popular pathways for the synthesis of isoflavones are the deoxybenzoin and the chalcone routes. In the first route, isoflavones are synthesized by the ring closure of deoxybenzoin with C1 unit by using different reagents such as ethoxalylchoride in pyridine [12] , triethylorthoformate with pyridine and piperidine [13] , N, N-dimethylformamide and BF3∙Et2O with MeSO2Cl [14] [15] or POCl3 [16] , anhydrous ethyl formate and powdered sodium [17] , acetic anhydride and sodium acetate [18] [19] , acetic-formic anhydride [20] , N-formylimidazole in anhydrous THF [21] and phenyliodine (III)bis (trifluoroacetate) [22] [23] . In the second route, isoflavones are synthesized by oxidative rearrangement of a chalcone using reagents like thallium(III)nitrate [24] -[29] and thallium(III)acetate [30] -[32] . Similarly, rearrangement of chalcone epoxide with BF3∙Et2O is followed by catalytic hydrogenation [33] [34] . Other routes include the conversion of flavanones into isoflavones by thallium(III)nitrate in a mixture of CH3OH and CHCl3 [35] , arylation of 4-chromanones with 4, 5-dimethoxy-o-benzoquinone in anhydrous DMSO followed by acidification [36] [37] , tetrakis (triphenylphosphine) palladium (0) catalyzed cross-coupling reactions of 3-iodo- chromone with arylboronic acid [38] , etc. However, the reported syntheses of many isoflavones including daidzein and formononetin are time-consuming (Bass, 1976 [44] , Baker et al., 1953 [12] , Farkas et al., 1971 [43] , Pelter and Foot, 1976 [50] , Yoder et al., 1954 [51] ).
Homoisoflavonoids are a class of naturally occurring oxygen containing heterocyclic compounds. Both natural and synthetic homoisoflavonoids exhibit numerous biological activities [39] -[41] like antifungal, hypocholesterolemic, antimutagenic, antirhinovirus, antiallergic, angio productive activity, antihistaminic activity, anti- inflammatory, antioxidant, antiviral, cough relief, inhibition of platelet aggregation etc. Homoisoflavonoids can be synthesized either by the condensation of 4-chromanones with arylaldehdes in methanol by passing HCl gas or by using piperidine as a base followed by isomerisation of the double bond using Pd/C at 250˚C [42] or by the extension of one carbon in dihydrochalcone using ethylformate/sodium [43] or BF3∙Et2O and DMF with MeSO2Cl [44] or PCl5 [45] etc.Both the methods have disadvantages; while the first method has multiple steps, in the second method, the phenolic hydroxyls have to be protected to get chalcones in good yield.
However, most of the methods reported for the synthesis of chromones, isoflavones and homoisoflavones suffer from harsh reaction conditions, poor substituent tolerance, long reaction times, and low to moderate yields. Therefore, developing a milder and more general procedure for chromones, isoflavones, and homoisoflavones is still highly desirable. It was reported that when DMF was treated with phthaloyl dichloride in 1,4-dioxane at 40˚C for 3 h precipitated only vilsmeier reagent as a solid form while the co-product phthalic anhydride was dissolved in a solvent [46] . The precipitates were collected by filtration through a glass-filter funnel under nitrogen atmosphere. The residue was dried in vacuo to give white crystals, which was identified as the vilsmeier reagent by comparison with authentic supplied by Aldrich Chemical Co. Vilsmeier reagent is well known as a versatile synthetic tool for the formylation of electron-rich aromatics, chlorination of alcohols, conversion of carboxylic acid into the corresponding acid chloride and so on [47] [48] . A series of 2-hydroxyacetophenone, deoxybenzoin and dihydrochalcone was cyclized with a one carbon unit by using this reagent. The reaction requires a short reaction time, mild reaction conditions and easy work-up. Products obtained by this methodology do not have contaminants such as sulphur or phosphine obtained through DMF with MeSO2Cl [44] or PCl5 [45] . Naturally, occurring isoflavones such as formononetin (9c), daidzein (9d) and retusin (9h) was synthesized by applying this methodology. To the best of researcher knowledge, the synthesis of chromones, isoflavones and homoisoflavones using vilsmeier reagent formed from phthaloyl dichloride and DMF has not been reported.
2. Results and Discussion
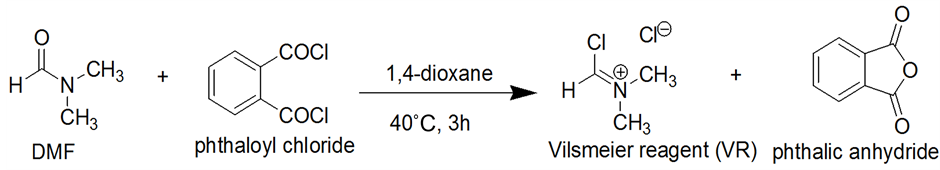
The method first involved the preparation of vilsmeier reagent, for this, to a mixture of DMF in 1,4-dioxane was added phthaloyl dichloride at room temperature, and then the whole mixture was stirred at 40˚C for 3 h (Scheme 1). The white precipitates of (chloromethylene) dimethyliminiumchloride (VR) were isolated by filtration under a nitrogen atmosphere.
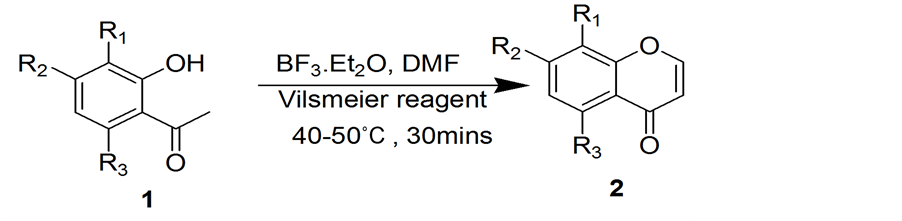
First application came with its usage in preparation of chromones in which 1 equiv. of substituted 2-hy- droxyacetophenone was dissolved in3 equiv. of BF3∙Et2O and DMF was added drop wise with stirring at 10˚C. Then whole reaction mixture was transferred slowly with continuous stirring into 1 equiv. of vilsmeier reagent. The reaction mixture was stirred at 50˚C for 30 minutes (Scheme 2). The completion of reaction was monitored by TLC. The reaction mixture was poured into 3N HCl, extracted with EtOAc, dried over Na2SO4 and concen-
Scheme 1. Preparation of vilsmeier reagent from phthaloyl dichloride and DMF.
Scheme 2. Synthesis of chromones.
trated. The pure compound was then harvested with column chromatography.
The next success came with its usage in the preparation of isoflavones and homoisoflavones. For this, deoxybenzoins (7) and dihydrochalcones (8) were prepared by the published procedure [49] from phenyl acetic acid and 3-phenylpropanoic acid respectively with substituted phenols by Friedel-Crafts acylation using BF3∙Et2O which served as the Lewis acid for the acylation as well as the solvent for the reaction. The acylation was carried out at 85˚C - 90˚C. The completion of reaction was monitored by TLC. In most cases, the reaction was completed within 90 minutes. However, the substitution pattern as well as the presence of unprotected hydroxyl groups on the aromatic rings influenced the reaction time and the product yield. Conversion of these intermediate deoxybenzoin and dihydrochalcone into their respective isoflavones and homoisoflavones can be carried out either directly by treating with vilsmeier reagent, a minimum of 5 equivalents of BF3∙Et2O was required (Method A) or these intermediates were isolated, purified and then cyclised with vilsmeier reagent, for this a minimum of 3 equivalents of BF3∙Et2O was required (Method B) (Scheme 3). In all cases, the reaction was completed in 30-40 mins and the products were characterized by their spectral data (IR, NMR, and mass spectrometry).
To explain the formation of chromones (2), isoflavones (9), and homoisoflavones (10), a suggested mechanism is shown in Figure 1. The mechanism involves the addition of a vilsmeier reagent (I) to the acetophenone. BF3 complex (II) to form (III), with subsequent nucleophilic attack of the hydroxyl group of 2-hydroxyaceto- phenone to form (IV), which is deaminated to form the final product (V).
3. Conclusion
A variety of chromones, isoflavones and homoisoflavones were synthesized in excellent yields using vilsmeier reagent generated from phthaloyl dichloride and DMF as the key reagent. The ready availability, low cost of phthaloyl dichloride and DMF, high activity of isolated vilsmeier reagent, the short reaction time, the mild reaction conditions, and the easy purification of the products make this an attractive new method for the synthesis of chromones, isoflavones and homoisoflavones, etc.
4. Experimental
4.1. General Remarks
All synthesized compound melting points were recorded on a Mel-Temp melting point apparatus in open capillaries and are uncorrected. Reactions requiring anhydrous conditions were performed in flame-dried glassware, and cooled under an argon or nitrogen atmosphere. Acme silica gel G and silica gel (100 - 200 mesh) were used for analytical thin-layer chromatography and column chromatography. Visualization of the resulting chroma-
![]()
Figure 1. Plausible mechanism for the formation of 2, 9, and 10.
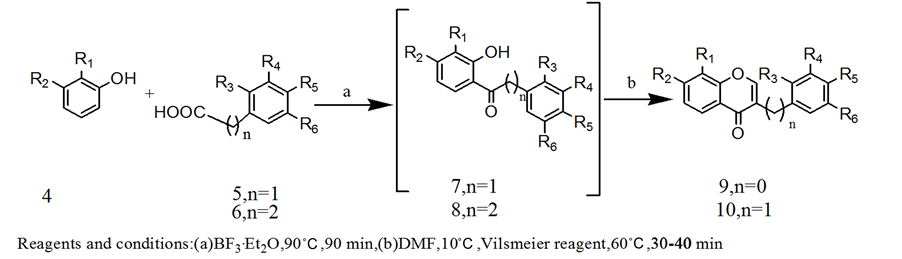
Scheme 3. Synthesis of isoflavones (9a-9h) and homoisoflavones (10a-10f).
tograms was done by looking under an ultraviolet lamp (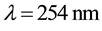 ). IR spectra were recorded on a Perkin-Elmer BX1 FTIR spectrophotometer and 1H NMR (400 MHz) and 13C NMR (100 MHz) spectra were recorded on a Bruker AMX 400 MHz. NMR spectrometer using TMS as the internal standard and the values for chemical shifts (_) being given in parts per million and coupling constants (J) in hertz. Mass spectra were recorded on an Agilent 1100 LC/MSD.
). IR spectra were recorded on a Perkin-Elmer BX1 FTIR spectrophotometer and 1H NMR (400 MHz) and 13C NMR (100 MHz) spectra were recorded on a Bruker AMX 400 MHz. NMR spectrometer using TMS as the internal standard and the values for chemical shifts (_) being given in parts per million and coupling constants (J) in hertz. Mass spectra were recorded on an Agilent 1100 LC/MSD.
4.2. Preparation of Vilsmeier Reagent from Phthaloyl Dichloride and DMF in 1,4-Dioxane
A mixture of DMF 30 g (0.41 mol) and phthaloyl dichloride 90 g (0.44 mol) in 1,4-dioxane (330 mL) was stirred at 40˚C for 3 h. The white precipitates of (chloromethylene)dimethyliminium chloride (VR) that formed were collected by filtration under a nitrogen atmosphere, washed with 1,4-dioxane (100 mL × 2) and hexane (100 mL), and dried under reduced pressure, 41 gm (78% yield).
4.3. Preparation of Chromones (2a-2d)
DMF (4.6 mL) was added to a stirred solution of 2-hydroxyacetophenone (3 mmol) in BF3∙Et2O (7.5 mmol) at 10˚C for 5 min. The reaction mixture was then added to the vilsmeier reagent (4.5 mmol) drop wise with stirring at room temperature. After completion of addition, the reaction mixture was stirred at 50˚C for 30 - 40 mins and poured into boiling dilute HCl slowly and cooled. The solution was extracted with ethyl acetate (30 mL × 2) and the combined organic layer was dried over anhydrous Na2SO4. The crude obtained after evaporation of the solvent was chromatographed over silica gel column using chloroform-methanol mixtures as eluent to give 2a-2d.
4.4. Chromen-4-One (2a) (Table 1, Entry 1)
Colorless solid; yield 376 mg (80%); mp 55˚C - 58˚C. 1H NMR (400 MHz, DMSO-d6)_: 6.32 (d, J = 5.6 Hz, 1H), 7.35 - 7.43 (m, 2H), 7.64 (t, J = 7.6 Hz, 1H), 7.86 (d, J = 5.6 Hz, 1H), 8.18 (d, J = 7.6 Hz, 1H). 13C NMR (100 MHz, DMSO-d6)_= 111.9, 117.2, 123.9, 124.3, 124.7, 132.8, 154.5, 155.5, 176.6. LC-MS: m/z: 147 [M +
![]()
Table 1. Synthesis of chromones (2a-2d).
1]+. Anal.calcd. for C9H6O2: C 73.97, H 4.14; Found: C 73.94, H 4.19.
4.5. 7-Hydroxy-4H-Chromen-4-One (2b) (Table 1, Entry 2)
Pale brown solid; yield 412 mg (85%); mp 206˚C - 208˚C. 1H NMR (400 MHz, DMSO-d6)_: 6.21 (d, J = 6.0 Hz, 1H), 6.84 (d, J = 2.4 Hz, 1H), 6.91 (dd, J = 2.4, 8.4 Hz, 1H), 7.87 (d, J = 8.4 Hz, 1H), 8.14 (d, J = 6.0 Hz, 1H), 10.76 (s, 1H). 13C NMR (100 MHz, DMSO-d6)_= 102.3, 111.9, 115.0, 117.0, 126.6, 156.0, 157.7, 162.5, 175.5. LC-MS: m/z: 161 [M-1]-. Anal.calcd. for C9H6O3: C 66.67, H 3.73; found: C 66.65, H 3.75.
4.6. 7,8-Dihydroxy-4H-Chromen-4-One (2c) (Table 1, Entry 3)
Brown solid; yield 466 mg (87%); mp 205˚C - 208˚C. 1H NMR (400 MHz, DMSO-d6)_: 6.17 (d, J = 6.0 Hz, 1H), 6.93 (d, J = 6.8 Hz, 1H), 7.37 (d, J = 6.8 Hz, 1H), 8.19 (d, J = 6.0 Hz, 1H), 9.40 (s, 1H), 10.29 (s, 1H). 13C NMR (100 MHz, DMSO-d6)_= 111.3, 114.0, 115.0, 117.8, 132.9, 146.9, 150.0, 155.7, 176.0. LC-MS: m/z: 177 [M-1]−. Anal.calcd. for C9H6O4: C 60.68, H 3.39; found: C 60.63, H 3.43.
4.7. 5,7-Dihydroxy-4H-Chromen-4-One (2d) (Table 1, Entry 4)
Brown solid; yield 474 mg (89%); mp 268˚C - 270˚C. 1H NMR (400 MHz, DMSO-d6)_: 6.20 (d, J = 2.0 Hz, 1H), 6.27 (d, J = 6.0 Hz, 1H), 6.36 (d, J = 2.0 Hz, 1H), 8.17 (d, J = 6.0 Hz, 1H), 10.85 (s, 1H), 12.69 (s, 1H). 13C NMR (100 MHz, DMSO-d6)_= 93.9, 98.9, 104.8, 110.4, 149.8, 157.3, 157.7, 164.2, 181.2. LC-MS: m/z: 177 [M-1]−. Anal.calcd. for C9H6O4: C 60.68, H 3.39; found: C 60.65, H 3.41.
5. General Experimental Procedure for Isoflavones (9a-9h)
5.1. Method A
A mixture of substituted phenol (3 mmol), phenylacetic acid (3 mmol), and BF3∙Et2O (15 mmol) was refluxed at 90˚C for 90 min under Nitrogen atmosphere. The reaction mixture was then cooled to 10˚C and DMF (4.6 mL) was added drop wise. The above reaction mixture was then added drop wise with stirring into vilsmeier reagent (4.5 mmol) at room temperature. After completion of addition, the reaction mixture was stirred at 60˚C for 30 - 40 min and poured into boiling dilute HCl slowly and cooled. The solution was extracted with ethylacetate (30 mL × 2) and the organic layer was dried over anhydrous Na2SO4. The crude obtained after evaporation of the solvent was chromatographed over a silica gel column using chloroform-methanol mixtures as eluent to give isoflavones (9a-9h).
5.2. Method B
A mixture of substituted phenol (3 mmol), phenylacetic acid (3 mmol), and BF3∙Et2O (9 mmol) was refluxed at 90˚C for 90 min under Nitrogen atmosphere. The mixture was then poured into NaOAc solution (100 mL, 10%) and allowed to stand for 4 hr and the solution was extracted with EtOAc (3 × 100 mL). The combined organic layer was washed with water (20 mL) and brine (20 mL) and dried over anhydrous Na2SO4. The crude obtained after evaporation of the solvent was chromatographed over a silica gel column using hexane-EtOAc mixtures as eluent to give deoxybenzoins (7f-7h). The purified materials were then used for the synthesis of isoflavones. A mixture of deoxybenzoin (3 mmol) and BF3∙Et2O (7.5 mmol) was cooled to 10˚C and DMF (4.6 mL) was added drop wise. The cyclization procedure and work up are similar to method A.
5.3. 7-Hydroxy-3-Phenyl-4H-Chromen-4-One (9a) (Table 2, Entry 1)
White solid; yield (method A) 627 mg (88%); mp 210˚C - 213˚C. 1H NMR (400 MHz, DMSO-d6)_: 6.88 (d, J = 2.4 Hz, 1H), 6.96 (dd, J = 2.4, 8.4 Hz, 1H), 7.34 - 7.44 (m, 3H), 7.57 (d, J = 7.2 Hz, 2H), 7.99 (d, J = 8.8 Hz, 1H), 8.36 (s, 1H), 10.80 (s, 1H). 13C NMR (100 MHz, DMSO-d6)_= 102.1, 115.2, 116.6, 123.5, 127.2, 127.6, 128.0, 128.8, 132.1, 153.6, 157.4, 162.6, 174.3. LC-MS: m/z: 237 [M-1]-. Anal.calcd. for C15H10O3: C 75.62, H 4.23; found: C 75.60, H 4.27.
5.4. 7-Hydroxy-3-(3-Methoxyphenyl)-4H-Chromen-4-One (9b) (Table 2, Entry 2)
Pale pink solid; yield (method A) 715 mg (89%); mp 215˚C - 217˚C. 1H NMR (400 MHz, DMSO-d6)_: 3.78 (s, 3H), 6.88 (s, 1H), 6.93 - 6.96 (m, 2H), 7.13 - 7.15 (m, 2 H), 7.33 (t, J = 8.0 Hz, 1H), 7.98 (d, J = 8.8 Hz, 1H), 8.38 (s, 1H), 10.79 (s, 1H) 13C NMR (100 MHz, DMSO-d6)_= 55.06, 102.1, 113.2, 114.6, 115.2, 116.6, 121.1, 123.3, 127.2, 129.0, 133.4, 153.8, 157.3, 159.0, 162.6, 174.3. LC-MS: m/z: 267 [M-1]-. Anal.calcd. for C16H12O4: C 71.64, H 4.51; found: C 71.60, H 4.56.
5.5. 7-Hydroxy-3-(4-Methoxyphenyl)-4H-Chromen-4-One (9c) (Table 2, Entry 3)
Off-white solid; yield (method A) 723 mg (90%); mp 257˚C - 258˚C. 1H NMR (400 MHz, DMSO-d6)_: 3.78 (s, 3H), 6.88 (d, J = 2.0 Hz, 1H), 6.94 (dd, J = 2.4, 8.8 Hz, 1H), 6.98 (d, J = 8.8 Hz, 2H), 7.49 (d, J = 8.8 Hz, 2H), 6.97 (d, J = 8.8 Hz, 1H), 8.31 (s, 1H). 13C NMR (100 MHz, DMSO-d6)_= 55.1, 102.0, 113.6, 114.1, 115.0, 116.6, 123.1, 124.1, 127.2, 130.0, 153.0, 157.4, 158.9, 162.3, 174.6. LC-MS: m/z: 267 [M-1]-. Anal.calcd. for C16H12O4: C 71.64, H 4.51; found: C 71.60, H 4.55.
5.6. 7-Hydroxy-3-(4-Hydroxyphenyl)-4H-Chromen-4-One (9d) (Table 2, Entry 4)
Pale brown powder; yield (method A) 670 mg (88%); mp 310˚C - 312˚C. 1H NMR (400 MHz, DMSO-d6)_: 6.79 (d, J = 8.4 Hz, 2H), 6.83 (d, J = 2.0 Hz, 1H), 6.91 (dd, J = 2.0, 8.0 Hz, 1H), 7.36 (d, J = 8.4 Hz, 2H), 7.96 (d, J = 8.0 Hz, 1H), 8.28 (s, 1H), 9.55 (s, 1H), 10.83 (s, 1H). 13C NMR (100 MHz, DMSO-d6)_= 102.0, 114.9, 115.1, 122.5, 123.5, 127.2, 130.0, 157.2, 157.3, 162.4, 174.7. LC-MS: m/z: 253 [M-1]-. Anal.calcd. for C15H10O4: C 70.86, H 3.96; found: C 70.85, H 3.98.
5.7. 7-Hydroxy-3-(2, 4-Dimethoxyphenyl)-4H-Chromen-4-One (9e) (Table 2, Entry 5)
Off white solid; yield (method A) 795 mg (89%); mp 265˚C - 270˚C. 1H NMR (400 MHz, DMSO-d6)_: 3.70 (s, 3H), 3.80 (s, 3H), 6.56 (d, J = 8.0 Hz, 1H), 6.63 (s, 1H), 6.86 (s, 1H), 6.93 (d, J = 8.8 Hz, 1H), 7.13 (d, J = 8.0 Hz, 1H), 7.92 (d, J = 8.8 Hz, 1H), 8.11 (s, 1H), 10.73 (s, 1H) 13C NMR (100 MHz, DMSO-d6)_= 55.2, 55.5, 98.6, 102.1, 104.6, 113.5, 114.9, 116.5, 121.5, 127.1, 132.0, 153.8, 157.4, 158.4, 160.6, 162.4, 174.3. LC-MS: m/z: 297 [M-1]−. Anal. calcd. for C17H14O5: C 68.45, H 4.73; found: C 68.46, H 4.75.
![]()
Table 2. Synthesis of isoflavones (9a-9h).
aUnoptimized condition; bDeoxybenzoins were isolated and converted into isoflavones.
5.8. 7,8-Dihydroxy-3-Phenyl-4H-Chromen-4-One (9f) (Table 2, Entry 6)
Pale brown solid; yield (method A) 571 mg (75%), yield (method B) 677 mg (89%); mp 200˚C - 205˚C. 1H NMR (400 MHz, DMSO-d6)_: 6.98 (d, J = 8.8 Hz, 1H), 7.37 - 7.44 (m, 3H), 7.49 (d, J = 8.8 Hz, 1H), 7.59-7.57 (m, 2H), 8.43 (s, 1H), 9.46 (s, 1H), 10.33 (s, 1H). 13C NMR (100 MHz, DMSO-d6)_= 114.2, 115.6, 117.4, 123.0, 127.5, 128.0, 128.9, 132.2, 132.9, 146.7, 150.1, 153.5, 174.8. LC-MS: m/z: 253 [M-1]−. Anal.calcd. for C15H10O4: C 70.86, H 3.96; found: C 70.84, H 4.00.
5.9. 7,8-Dihydroxy-3-(3-Methoxyphenyl)-4H-Chromen-4-One (9g) (Table 2, Entry 7)
Brown solid; yield (method A) 597 mg (70%), yield (method B) 725 mg (85%); mp 216˚C - 218˚C. 1H NMR (400 MHz, DMSO-d6)_: 3.78 (s, 3H), 6.94 (d, J = 7.6 Hz, 1H), 6.97 (d, J = 8.8 Hz, 1H), 7.14 - 7.17 (m, 2H), 7.33 (t, J = 7.6 Hz, 1H), 7.49 (d, J = 8.8 Hz, 1H), 8.44 (s, 1H) 9.41 (s, 1H), 10.32 (s, 1H). 13C NMR (100 MHz, DMSO-d6)_= 55.0, 113.1, 114.2, 114.7 115.7, 117.4, 121.2, 122.7, 129.0, 132.9, 133.5, 146.6, 150.1, 153.6, 158.9, 174.7. LC-MS: m/z: 283 [M-1]−. Anal. calcd. for C16H12O5: C 67.60, H 4.25; found: C 67.58, H 4.28.
5.10. 7,8-Dihydroxy-3-(4-Methoxyphenyl)-4H-Chromen-4-One (9h) (Table 2, Entry 8)
Pale Brown solid; yield (method A) 640 mg (75%), yield (method B) 768 mg (90%); mp 252˚C - 254˚C. 1H NMR (400 MHz, DMSO-d6)_: 3.79 (s, 3H), 6.96 (d, J = 8.8 Hz, 1H), 6.99 (d, J = 8.8 Hz, 2H), 7.48 (d, J = 8.8 Hz, 1H), 7.52 (d, J = 8.8 Hz, 2H), 8.38 (s, 1H), 9.42 (s, 1H), 10.29 (s, 1H). 13C NMR (100 MHz, DMSO-d6)_= 55.1, 113.5, 114.1, 115.6, 117.4, 122.6, 124.4, 130.0, 132.8, 146.7, 149.8, 150.0, 152.8, 158.9, 175.0. LC-MS: m/z: 283 [M-1]−. Anal. calcd. for C16H12O5: C 67.60, H 4.25; found: C 67.59, H 4.28.
6. General Experimental Procedure for Homo-Isoflavones (10a-10f)
6.1. Method A
A mixture of substituted phenol (3 mmol), 3-phenylpropanoic acid (3 mmol), and BF3∙Et2O (15 mmol) was refluxed at 90˚C for 90 min under Nitrogen atmosphere. The reaction mixture was then cooled to 10˚C and DMF (4.6 mL) was added drop wise. The above reaction mixture was then added drop wise with stirring into vilsmeier reagent (4.5 mmol) at room temperature. After completion of addition, the reaction mixture was stirred at 60˚C for 30 - 40 min and poured into boiling dilute HCl slowly and cooled. The solution was extracted with ethylacetate (30 mL × 2) and the organic layer was dried over anhydrous Na2SO4. The crude obtained after evaporation of the solvent was chromatographed over a silica gel column using chloroform-methanol mixtures as eluent to give homo-isoflavones (10a-10h).
6.2. Method B
A mixture of substituted phenol (3 mmol), 3-phenylpropanoic acid (3 mmol), and BF3∙Et2O (9 mmol) was refluxed at 90˚C for 90 min under Nitrogen atmosphere. The mixture was then poured into NaOAc solution (100 mL, 10%) and allowed to stand for 4 hr and the solution was extracted with EtOAc (3 × 100 mL). The combined organic layer was washed with water (20 mL) and brine (20 mL) and dried over anhydrous Na2SO4. The crude obtained after evaporation of the solvent was chromatographed over a silica gel column using hexane-EtOAc mixtures as eluent to give dihydrchalcones (8d-8f). The purified materials were then used for the synthesis of homoisoflavones. A mixture of dihydrochalcone (3 mmol) and BF3∙Et2O (7.5 mmol) was cooled to 10˚C and DMF (4.6 mL) was added drop wise. The cyclization procedure and work up are similar to method A.
6.3. 3-Benzyl-7-Hydroxy-4H-Chromen-4-One (10a) (Table 3, Entry 1)
Pale pink solid; yield (method A) 635 mg (84%); mp 210˚C - 214˚C. 1H NMR (400 MHz, DMSO-d6)_: 3.67 (s, 2H), 6.82 (d, J = 2.4 Hz, 1H), 6.89 (dd, J = 2.4, 8.8 Hz, 1H), 7.14 - 7.18 (1H), 7.23 - 7.29 (m, 4H), 7.87 (d, J = 8.8 Hz, 1H), 8.17 (s, 1H), 10.72 (s, 1H). 13C NMR (100 MHz, DMSO-d6)_= 30.6, 102.1, 114.9, 116.2, 122.8, 125.9, 126.7, 128.1, 128.4, 139.7, 153.2, 157.7, 162.4, 175.4. LC-MS: m/z: 251 [M-1]−. Anal. calcd. for C16H12O3: C 76.18, H 4.79; found: C 76.14, H 4.82.
![]()
Table 3. Synthesis of homoisoflavones (10a-10f).
aUnoptimized condition. bDihydrochalcones were isolated and converted into homoisoflavones.
6.4. 7-Hydroxy-3-(4-Methoxybenzyl)-4H-Chromen-4-One (10b) (Table 3, Entry2)
Pale brown solid; yield (method A) 736 mg (87%); mp 161˚C - 165˚C. 1H NMR (400 MHz, DMSO-d6)_: 3.59 (s, 2H), 3.69 (s, 3H), 6.81 - 6.83 (m, 3H), 6.89 (dd, J = 2.0, 8.8 Hz, 1H), 7.20 (d, J = 8.4 Hz, 2H), 7.87 (d, J = 8.8 Hz, 1H), 8.12 (s, 1H), 10.71 (s, 1H). 13C NMR (100 MHz, DMSO-d6)_= 29.8, 54.9, 102.1, 113.6, 114.9, 116.2, 123.3, 126.7, 129.0, 129.5, 131.5, 153.0, 157.6, 157.7, 162.4, 175.4. LC-MS: m/z: 281 [M-1]-. Anal.calcd. for C17H14O4: C 72.33, H 5.00; found: C 72.30, H 5.05.
6.5. 7-Hydroxy-3-(4-Hydroxybenzyl)-4H-Chromen-4-One (10c) (Table 3, Entry3)
Colorless solid; yield (method A) 715 mg (89%); mp 210˚C - 212˚C. 1H NMR (400 MHz, DMSO-d6)_: 3.52 (s, 2H), 6.64 (d, J = 8.4 Hz, 2H), 6.79 (d, J = 2.0 Hz, 1H), 6.89 (dd, J = 8.8, 2.0 Hz, 1H), 7.07 (d, J = 8.4 Hz, 2H), 7.86 (d, J = 8.8 Hz, 1H), 8.05 (s, 1H), 9.85 (s, 1H), 10.75 (s, 1H) 13C NMR (100 MHz, DMSO-d6)_= 30.2, 102.1, 114.5, 115.1, 116.3, 123.9, 126.7, 128.9, 129.4, 152.3, 155.5, 157.8, 162.3, 176.3. LC-MS: m/z: 267 [M-1]. Anal.calcd. for C16H12O4: C 71.64, H 4.51; found: C 71.62, H 4.54.
6.6. 7-Hydroxy-3-(2, 5-Dimethoxybenzyl)-4H-Chromen-4-One (10d) (Table 3, Entry4)
Light brown solid; yield (method A) 823 mg (88%); mp 184˚C - 188˚C. 1H NMR (400 MHz, DMSO-d6)_: 3.59 (s, 2H), 3.69 (s, 3H), 3.75 (s, 3H), 6.70 - 6.75 (m, 2H), 6.80 (d, J = 2.0 Hz, 1H), ), 6.85 (d, J = 8.8 Hz, 1H), 6.90 (dd, J = 2.0, 8.8 Hz, 1H), 7.88 (s, 1H), 7.89 (d, J = 8.8 Hz, 1H), 10.68 (s, 1H). 13C NMR (100 MHz, DMSO- d6)_= 25.2, 55.2, 55.7, 102.1, 111.3, 111.6, 114.9, 116.1, 116.4, 121.7, 126.7, 128.0, 151.1, 151.2, 152.9, 153.2, 157.7, 162.4, 175.4. LC-MS: m/z: 311 [M-1]−. Anal.calcd. for C18H16O5: C 69.22, H 5.16; found: C 69.19, H 5.19.
6.7. 7,8-Dihydroxy-3-(4-Methoxybenzyl)-4H-Chromen-4-One (10e) (Table 3, Entry 5)
Colorless solid; yield (method A) 698 mg (78%), yield (method B) 785 mg (88%); mp 250˚C - 253˚C. 1H NMR (400 MHz, DMSO-d6)_: 3.60 (s, 2H), 3.68 (s, 3H), 6.81 (d, J = 8.4 Hz, 2H), 6.93 (d, J = 8.8 Hz, 1H), 7.20 (d, J = 8.4 Hz, 2H), 7.39 (d, J = 8.8 Hz, 1H), 8.19 (s, 1H), 9.34 (s, 1H), 10.20 (s, 1H). 13C NMR (100 MHz, DMSO-d6)_ = 29.8, 54.9, 113.6, 114.0, 115.1, 117.1, 122.7, 129.4, 131.6, 132.8, 147.0, 149.8, 152.8, 157.6, 175.9. LC-MS: m/z: 297 [M-1]−. Anal.calcd. for C17H14O5: C 68.45, H 4.73; found: C 68.42, H 4.79.
6.8. 7,8-Dihydroxy-3-(2, 5-Dimethoxybenzyl)-4H-Chromen-4-One (10f) (Table 3, Entry 6)
Pale brown solid; yield (method A) 786 mg (80%), yield (method B) 836 mg (85%); mp 226˚C - 230˚C. 1H NMR (400 MHz, DMSO-d6)_: 3.60 (s, 2H), 3.64 (s, 3H), 3.75 (s, 3H), 6.72 - 6.75 (m, 2H), 6.88 (d, J = 8.4 Hz, 1H), 6.93 (d, J = 8.8 Hz, 1H), 7.39 (d, J = 8.8 Hz, 1H), 7.98 (s, 1H), 9.32 (s, 1H), 10.20 (s, 1H). 13C NMR (100 MHz, DMSO-d6)_ = 25.2, 55.2, 55.8, 111.3, 111.5, 114.0, 115.1, 116.4, 116.9, 121.1, 128.1, 132.8, 147.0, 149.8, 151.2, 152.9, 153.0, 175.9. LC-MS: m/z: 327 [M-1]−. Anal. calcd. For C18H16O6: C 65.85, H 4.91; found: C 65.81, H 4.96.
Acknowledgements
The author is grateful to the Department of Organic Chemistry & FDW, Andhra University, Visakhapatnam, India for giving him the opportunity to pursue his PhD.