American Journal of Analytical Chemistry
Vol. 3 No. 4 (2012) , Article ID: 18439 , 10 pages DOI:10.4236/ajac.2012.34045
New Method for Optimization and Simultaneous Determination of Sparfloxacin and Non Steroidal Anti-Inflammatory Drugs: Its In-Vitro Application
1Faculty of Pharmacy, Jinnah University for Women, Karachi, Pakistan
2United Biotech (Pvt) Ltd., Karachi, Pakistan
3College of Pharmacy, Dow University of Health Sciences, Karachi, Pakistan
Email: drsomi1983@yahoo.com
Received February 24, 2012; revised March 24, 2012; accepted April 4, 2012
Keywords: Sparfloxacin; NSAIDs; HPLC; Interactions; ANOVA
ABSTRACT
A simple reversed phase HPLC method was developed and validated for the simultaneous determination of sparfloxacin (SPFX), diclofenac sodium, meloxicam, ibuprofen, flurbiprofen, naproxen and mefenemic acid in a relatively short time with high linearity in bulk material, pharmaceutical formulations and human serum. Purospher STAR C18 (250 × 4.6 mm, 5 mm) column was utilized with mobile phase, methanol and water (90:10, v/v pH 2.70 adjusted by phosphoric acid), was delivered at a flow rate of 1.5 mL∙min–1. Eluent was monitored using UV detector at 240 nm. The proposed method is specific, accurate (98.42% - 102.75%), precise (intra-day and inter-day variation 0.011% - 1.85%) and linear (R2 > 0.999) with in the desired range 0.15 - 40 µg∙mL–1 and the detection and quantification limit was 1.19E+08 – 0.150 µg∙mL–1 and 3.62E+08 – 0.4574 µg∙mL–1 respectively for SPFX and NSAIDs. The analysis of variance (ANOVA) and student’s t-test were applied to verify the results. The anticipated method is applicable to routine analysis of SPFX and NSAIDs in pharmaceutical formulations as well as in human serum samples. It has also applied on interaction of SPFX with NSAIDs.
1. Introduction
Sparfloxacin or cis-5-amino-1-cyclopropyl-7-(3,5-dimethylpiperazin-1-yl)-6,8-difluoro-1,4-dihydro-4-oxoquinoline-3-carboxylic acid (SPFX) (Figure 1) is an orally active synthetically broad spectrum third generation quinolone, characterized by good to excellent activity against Gram positive cocci (notably S. pneumoniae) and in selected agent activity against anaerobes and atypical pathogens. It is also moderately active against some (B. fragilis group) L. mono-cytogenes resistant [1-9] and has been shown to have excellent activity, not only against Neisseria gonorrhoeae, but also against Chlamydia trachomatis, Mycoplasmas and Gardnerella vaginalis [10].
Literature search assembled a number of different methods, which have been used for analysis of fluroquinolones in bulk, human serum and in pharmaceutical preparations. Gonzalez et al. [11] discovered the simultaneous determination of cefepime and the quinolones garenoxacin, moxifloxacin and levofloxacin in human urine by HPLC-UV. The method was applied to the determination of the four molecules in spiked samples of human urine. Nguyen et al. [12] reported simultaneous determination of levofloxacin, gatifloxacin and moxifloxacin in human serum. Nemutlue et al. [13] published simultaneous separation and determination of seven quinolones using HPLC and analysis of levofloxacin. Sultana et al. [14-16] provided simultaneous determination of NSAIDs with diltiazem and ceftrioxazone sodium. Koichi Suenami et al. [17] developed more sophisticated and sensitive analytical method for the simultaneous determination of NSAID’s in human plasma using Oasis HLB solid-phase extraction, followed by reversed-phase high-performance liquid chromatography and quadruple mass spectrometry with electrospray ionization operated in the negative ion mode. Koichi Suenami et al., [18] also
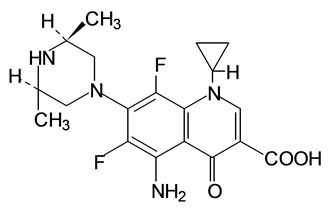
Figure 1. Structure of SPFX.
described a capillary liquid chromatography (LC) and quadruple mass spectrometry with electrospray ionization operated in the negative ion mode for the simultaneous determination of 16 NSAIDs in human plasma. Nagoji and friends [19] developed a simple, selective, rapid, precise and economical reverse phase HPLC method for simultaneous estimation of nimesulide and diclofenac sodium from capsules. There are few methods reported for the simultaneous determination of sparfloxacin with other drugs. Srinivas et al. [20] described HPLC method for simultaneous determination of sparfloxacin, moxifloxacin and gatifloxacin using levofloxacin as internal standard in human plasma. Degradation products studies by HPLC have been carried out by Marona et al. [21]. Akram et al. developed fluorescence probe enhanced spectrofluorimetric method for the determination of sparfloxacin in tablets and biological fluids and spectrophotometric determination method of sparfloxacin in pharmaceutical preparations by ternary complex formation with Pd (II) and eosin [22,23]. Young et al. [24] developed an HPLC method for quantitation of SPFX in human serum while. Argekar et al., Marona et al. and Nurun et al., developed methods for marketed products and stability testing [25-27]. Srikar [28] and friends developed spectrophotometric methods for quantitative estimation of sparfloxacin in bulk and pharmaceutical dosage forms.
Here, we report a simple, easy, quick and inexpensive isocratic RP-HPLC method with ultraviolet detection at 240 nm for the determination of SPFX in bulk, dosage form as well as in human serum. Low LOD and LOQ values also worth this method for the determination of sparfloxacin in clinical samples. Moreover, this method is applicable for “in-vitro” interaction studies of sparfloxacin with selected NSAIDs in a wide range of pH medium. Usually fluoroquinolones are prescribed for many diseases including respiratory and urinary tract infections. Historical background reveals that parallel administration of NSAIDs and new quinolones (NQ) can induce a synergistic interaction that results in convulsions and NQinduced neurotoxic effect synergistically increased in the presence of NSAIDs [29-31]. For the same purpose, to determine the effect of simultaneous administration of sparfloxacin (quinolones) and NSAIDs, a new RP-HPLC method has been developed which is simple, fast, cheap and easy to perform. The method is equally valid for the determination in bulk materials, pharmaceutical dosage formulations and human serum and is no where else reported before.
2. Experimental
2.1. Reagents
Standard bulk drug sample of sparfloxacin was gifted by Abbott Laboratories (Pakistan) Ltd. HPLC grade methanol was obtained from Merck Schuchardt OHG, Darmstadt, Germany and Tedia company, INC. (USA). While the NSAIDs used were diclofenac sodium (Fenac 50 mg tablet), flurbiprofen (Vobifen 100 mg tablet), meloxicam (Xobix 7.5 mg tablets), mefenamic acid (Ponstan 250 mg tablet ), ibuprofen (Brufen 200 mg tablet) and naproxen (Proxen 250 mg tablet) of Tabros Pharma (Pakistan), Amson Vaccines and Pharma (Pvt) Ltd., Hillton Pharma (Pvt) Ltd., Parke Davis and Co. Ltd., Aventis (Pvt.) Ltd., Abbott Laboratories (Pakistan ) Ltd. and Roche Pakistan Ltd., respectively. Each product was labeled and expiry dates were not earlier than two years, at the time of study.
2.2. Instrumentation
Shimadzu HPLC system equipped with LC-20 AT VP Pump was utilized in the method development, SPD- 20AV Shimadzu UV visible detectors and Purospher STAR C18 (250 × 4.6 mm, 5 mm) column used for separation. The chromatographic and integrated data were recorded using a CBM-102 communication Bus Module Shimadzu to Intel Pentium 4 machine with Shimadzu CLASS-GC 10 software. Mobile phase was sonicated by DGU-14 AM on-line degasser, and filtered through 0.45- micron membrane filter. Rheodyne manual injector fitted with a 20 μL loop. Calibrated Pyrex glassware was used for the solution and mobile phase preparation.
2.3. Preparation of Solutions
10 mg of the SPFX and NSAIDs were diluted to 100 mL with mobile phase to produce a concentration of 100 μg∙mL−1. Working solutions of SPFX and NSAIDs were prepared by diluting the stock solutions of with the same solvent to contain 0.15 - 80 µg∙mL−1.
2.4. Analysis of Formulations
For quality control (QC) samples, twenty tablets of each formulation were accurately weighed, crushed to make a fine powder. Calculated amount of powder was weighed and found to be equivalent to 10 mg and transferred to a separate 100 mL volumetric flask. It was dissolved in the mobile phase. Solutions with high, medium and low concentrations i.e.; 80, 100 and 120% were prepared, and then filtered through a 0.45-μm Millipore filter, in order to separate out the insoluble excipients by the same procedure as calibration standards but using different stock solutions. The sample solution was suitably diluted and used for the analysis.
All these solutions, calibration and QC samples were stored at 20˚C. Once prepared, analyzed daily for inter and intra-day variations of the method.
2.5. Procedure for Human Serum
Plasma sample, obtained from healthy volunteers, was collected and stored at −20˚C. To an aliquot of 1.0 mL plasma, 10 mL of acetonitrile was added and the mixture was vortexed for one minute centrifuged at 10,000 rpm for 10 minutes. It was then alienated supernatant by filtration (0.45 µ pore size membrane filter). An aliquot serum sample was fortified with sparfloxacin and NSAIDs to get the final concentrations of 0.15 - 80 μg∙mL−1.
2.6. Procedure of Interaction Studies
Sparfloxacin and interacting drugs (NSAIDs solutions (100 μg∙mL–1) were prepared in buffers of pH 4, 7.4 and 9 individually. These solutions were mixed in 1:1 ratio in Erlenmeyer flasks individually to get 50 mg∙mL–1 concentrations; heated on a water bath at 37˚C ± 5˚C with constant stirring at 100 rpm speed. Aliquots of 5 mL were with-drawn at an interval 30 min for 180 minutes. The sample was diluted in suitable dilution then filtered through 0.45 m filter membrane. Three replicates of filtered samples were injected to HPLC system.
The concentration of each drug was determined using linear equation and % availability was calculated. Effect of pH on the availability of sparfloxacin in presence of interacting drugs, was also monitored.
2.7. Statistical Analysis
Standard regression curve analysis was performed by use of STATISTICA version 7.0 (USA), without intersecting through zero. Linearity graphs were obtained by Microsoft Excel 2007 software. SPSS software version 10.0 (Carry, NC, USA) was used for the calculation of means, standard deviations, homoscedasticity of the calibration plots, and Student’s t-test.
3. Results and Discussion
3.1 Method Development and Optimization
The aim of the present study was to develop a simple, isocratic, accurate and sensitive HPLC method for the simultaneous determination of SPFX and NSAIDs. Different C18 columns were used but SPFX and NSAIDs could not separate properly. Finally Purospher STAR C18 (25 × 4.6 mm, 5 μm) column provided the best peak shapes and efficiencies. To investigate the appropriate wavelength for simultaneous determination of sparfloxacin and all the NSAIDs, solutions of these compounds in the mobile phase were scanned by UV-visible spectrophotometer in the range 200 - 400 nm. From the overlaid UV spectra (Figure 2), it was observed there was no interference from the mobile phase or baseline disturbance at 240 nm. It was, therefore, concluded that 240 nm is the most appropriate wavelength for analysis with suitable sensitivity.
Chromatographic conditions, especially the composition of the mobile phase, were optimized through several trials to achieve symmetric peak shapes for SPFX and NSAIDs as well as shorter run time. Initially various mobile phases were tested to obtain the best separation and resolution. It was found that a mobile phase containing a certain proportion of methanol and water gave symmetric peak shapes for all drugs. A mobile phase containing high proportion of methanol gives shorter run time. Therefore the final mobile phase of methanol and water in the ratio of 90:10 (v/v), provided good resolution. These solvents are easily available, non-toxic, cheap and commonly used solvents for RP-HPLC. To select the optimum mobile phase pH range, 2.5 to 4.0 were investigated, excellent performance was achieved at pH 2.7 adjusted with phosphoric acid. Flow rate was 1.5 mL∙min–1 with isocratic elution.

Figure 2. Uv-visible spectra of sparfloxacin and NSAIDs.
3.2. Method Validation
The developed method was validated according to ICH guidelines [32] and USP 2002 [33]. It includes various parameters for example system suitability, selectivity, specificity, linearity, accuracy test, precision, robustness, ruggedness, sensitivity, limit of detection and quantification.
3.2.1. System Suitability
It is a vital section of method validation to make certain that the operational system is running appropriately throughout the analysis. The system was equilibrated with the initial mobile phase composition, followed by 10 injections of the same standard. These 10 consecutive injections were used to evaluate the system suitability on each day of method validation. Parameters of system suitability are peaks symmetry (symmetry factor), theoretical plates of the column, resolution, mass distribution ratio (capacity factor) and relative retention [32,34]. They are summarized in Table 1.
3.2.2 Linearity
The reason of the check for linearity was to demonstrate that the entire analytical system (including detector and data acquisition) presents a linear response and it is directly proportional over the relevant concentration range of analytes [34]. These curves were obtained using the linear least squares regression procedure as shown in Table 2 over the concentrations ranging from 0.15 - 80 μg∙mL–1. These analysis results reveals good linear correlations between all the drugs having correlation coefficient (r2) value >0.999. The homoscedasticity of the calibration plots, tested by Friedman’s tests were found to be significantly linear over the tested ranges (Figures 3(a) and (b)).
3.2.3. Accuracy
The accuracy of the method was determined from the recovery results of spiked placebo samples. Stock solutions of drugs in appropriate portions were spiked into blank placebo matrix to produce concentrations of 80%, 100% and 120% of the theoretical concentration. Mean recovery of spiked samples were in the ranges of 98.42% - 102.75%, Recovery tests were performed by adding known amounts of standard solutions to sample followed by analysis using proposed method. Five runs were performed for every concentration and then peak area was calculated (Table 3, Figure 3(c)). The average recovery for each level was calculated as indicated by Association of Official Analytical Chemists International [35-37]. Thus, it can be concluded that used excepients (normally present in tablets) did not interfere with drug present in tablets and that filtration medium did not absorb the drug

Table 1. System suitability parameters.

Table 2. Regression characteristics.

Table 3. Accuracy of sparfloxacin and NSAIDs.
 (a)
(a) (b)
(b)
Figure 3. (a) Representative chromatogram of SPFX and NSAIDs at 240 nm; (b) Representative chromatogram of SPFX and NSAIDs at 240 nm; (c) Representative chromatogram of SPFX and NSAIDs at 240 nm in both mobile phase and human serum; (d) Representative chromatogram of SPFX and NSAIDs at 240 nm with and without excepients.
to any extent. High recovery indicated that the method has a high degree of accuracy for the simultaneous determination of sparfloxacin with selected NSAIDs.
3.2.4. Precision
Precision of the anticipated method was determined by repeatability (intra-day precision) and intermediate precision (intra-day precision). It is expressed as relative standard deviation (RSD). Every sample was injected five times. Both intraand inter-day RSD values were in the range of 0.112% to 2.77% confirming good precision (Table 4). Student t-test was applied to evaluate the difference in results by variations. The t-stat value was lower than the t-two-tailed value indicating no significant difference between intra-day and inter-day precision (df = 4). The results were insignificant indicating no remarkable difference in intra-day and inter-day precision.


Table 4. Precision of sparfloxacin and NSAIDs (n = 5).
3.2.5. Limit of Detection (LOD) and Quantification (LOQ)
The LOD and LOQ are calculated as: LOD = 3.3 µ/S and LOQ =10 µ/S; where µ is the standard deviation of the lowest standard concentration and S is the slope of the standard curve. The limits of detection (LOD) and quantification (LOQ) were determined from the calibration curve. The LOD and LOQ were shown in Table 2.
3.2.6. Specificity and Selectivity
The selectivity and specificity of the method was documented by studying resolution factor of the peak of sparfloxacin from that of NSAID’s. The method confirmed good resolutions ≥2 (Table 1) and was found to be free of hindrance from the excepients used in pharmaceutical formulation, which indicated the specificity of the system (Figure 3(d)).
3.2.7. Ruggedness
The ruggedness of the method was established by determining SPFX and NSAIDs, in bulk materials, dosage formulation and in human serum in two different labs. Lab 1 was Research Institute of Pharmaceutical Sciences, Department of Pharmaceutical Chemistry, Faculty of Pharmacy, University of Karachi while other lab was lab 9, Department of Chemistry, Faculty of Science, University of Karachi. Two different instruments of same configuretion i.e. LC 20, were used on different days by different analytes (Table 4). All the results were in good limits.
3.2.8. Robustness
Robustness of the method was accomplished by intended modifications made to the method parameters such as composition, flow rate, pH of the mobile phase, injection volume and column temperature and was found quite stable indicated better robustness of the developed method. Results are shown in Table 5.
4. Application of the Proposed Method for In-Vitro Interaction Study
Simultaneous determination of sparfloxacin, naproxen, flurbiprofen, ibuprofen, mefenamic acid, diclofenac sodium, and meloxicam was achieved as above. The developed method was also used on spiking the analytes in human serum. The applicability of the proposed method was demonstrated for “in-vitro” interaction studies of sparfloxacin with these selected NSAIDs in simulated gastric juice and buffers of pH 4, 7.4 and 9. Sparfloxacin and the reacting drugs were analyzed by measuring the area under curve (AUC), % recovery and considerable drift in retention time. The results of possible interactions are given in Table 6.


Table 5. Robustness of the proposed method (n = 6).
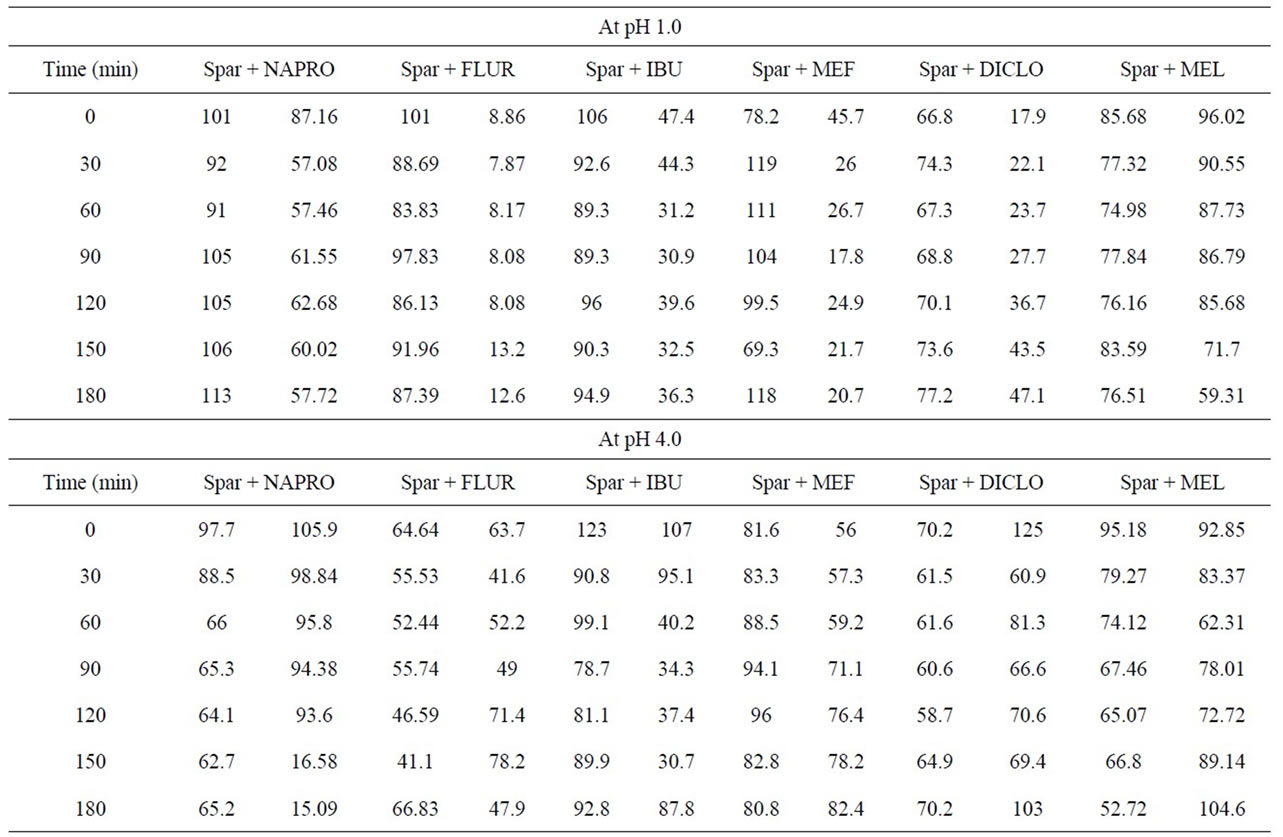

Table 6. % Recovery of sparfloxacin with NSAID’s.
Availability of naproxen decreased up to 57.72%, 15.09%, 66.22% and 59.77% in simulated gastric juice and buffers of pH 4, 7.4 and 9 respectively. Good interaction found between SPFX and flurbiprofen in simulated gastric juice (12.6%) and buffer of pH 4 (47.9%) while 64.8% and 62.7% available at pH 7.4 and 9 respectively. When ibuprofen interacted with sparfloxacin, the availability was significantly decreased up to 31.2% - 36.3% in selected simulated gastric juice and pH 7.4. While in pH 4 and 9, availability decreased to little extend. Availability of mefenamic acid significantly decreased in simulated gastric juice only. However, no or little interaction have been seen incase of pH 4, 7.4 and 9. The % availability of diclofenac Na decreased to 47.1 in simulated gastric juice, 14.5 in pH 7.4 and increased to 103% in pH 4 and 82.4 in pH 9. Meloxicam was available up to 59.31% - 104.6% at all preferred pH.
On the basis of above consequences, it can be concluded that these interactions were pH dependent and temperature also favored. Significant changes in % availability of drugs might be due to some changes in drug at its chromophoric group, resulting in the distinction of molar absorptivity value which itself is an evidence of drug interaction with NSAID’s. Moreover, variation in availability had occurred due to an addition of functional group being attached to the pharmacophore of sparfloxacin and due to loss of axuochromes. Advance in-vivo studies are needed for further evaluation.
5. Conclusion
A new method for the simultaneous determination of SPFX and NSAIDs, in bulk materials, pharmaceutical dosage formulation and human serum has been developed for the first time. The proposed HPLC method is simple, rapid, isocratic, specific, accurate and precise. Hence, it can be recommended for the routine quality control and evaluation of clinical data as well, of these drugs. It was apparent that sparfloxacin may interact with commonly used NSAID’s like ibuprofen, diclofenac sodium, flurbiprofen, Mefenamic acid and meloxicam which may result in convulsions. Therefore, above mentioned method is applicable in finding out the “in vitro” interactions of sparfloxacin with commonly used NSAID’s.
6. Acknowledgements
Authors wish to thank Higher Education Commission, Pakistan, for providing scholarship to Ms Somia Gul under Indigenous 5000 Ph.D Fellowship Program Batch IV.
REFERENCES
- J. M. Nelson, T. M. Chiller, J. H. Powers and F. J. Angulo, “Fluoroquinolone-Resistant Campylobacter Species and the Withdrawal of Fluoroquinolones from Use in Poultry: A Public Health Success Story,” Clinical Infectious Diseases, Vol. 44, No. 7, 2007, pp. 977-980. doi:10.1086/512369
- D. V. Ivanov and S. V. Budanov, “Ciprofloxacin and Antibacterial Therapy of Respiratory Tract Infections,” Antibiotiki i Khimioterapiia, Vol. 51, No. 5, 2006, pp. 29-37.
- A. Sivalakshmidevi, K. Vyas and G. Om Reddy, “Sparfloxacin, an Antibacterial Drug,” Acta Crystallographica, Vol. 56, No. 3, 2000, pp. 115-116.
- O. G. Francis, G. F. Roger and G. W. David, “Antibiotic and Chemotherapy,” 7th Edition, Churchill Livingstone, Philadelphia, 1997.
- Medical Economics Staff, “Physician Desk Reference,” 52nd Edition, Churchill Livingstone, New York, 1998.
- M. I. Andersson and A. P. MacGowan, “Development of the Quinolones,” Journal of Antimicrobial Chemotherapy, Vol. 51, Suppl. 1, 2003, pp. 1-11.
- M. S. Barrett, R. N. Jones, M. E. Erwin, D. M. Johnson and B. M. Briggs, “Antimicrobial Activity Evaluations of Two New Quinolones,” Diagnosis of Microbiology Infectious Diseases, Vol. 14, No. 5, 1991, pp. 389-401. doi:10.1016/0732-8893(91)90066-O
- G. C. Crumplin, “Aspects of Chemistry in the Development of the 4-Quinolone Antibacterial Agents,” Reviews of Infectious Diseases, Vol. 10, Suppl. 1, 1988, pp. 2-9.
- N. Sultana, M. S. Arayne, S. Gul and S. Shahmim, “Sparfloxacin-Metal Complexes as Antifungal Agents—Their Synthesis, Characterization and Antimicrobial Activities,” Journal of Molecular Structure, Vol. 975, No. 1-3, 2010, pp. 285-291. doi:10.1016/j.molstruc.2010.04.038
- P. H. Edelstein, M. A. C. Edelstein, K. H. Lehr and J. J. Ren, “In-Vitro Activity of Levofloxacin against Clinical Isolates of Legionella spp., Its Pharmacokinetics in Guinea Pigs, and Use in Experimental Legionella pneumophila Pneumonia,” Journal of Antimicrobial Chemotherapy, Vol. 37, No. 1, 1996, pp. 117-126. doi:10.1093/jac/37.1.117
- J. A. O. Gonzalez, M. C. Mochon and F. J. B. De La Rosa, “Simultaneous Determination of Cefepime and the Quinolones Garenoxacin, Moxifloxacin and Levofloxacin in Human Urine by HPLC-UV,” Mikrochimica Acta, Vol. 151, No. 1-2, 2005, pp. 39-45. doi:10.1007/s00604-005-0391-y
- H. A. Nguyen, J. Grellet, B. B. Ba, C. Quentin and M. C. Saux, “Simultaneous Determination of Levofloxacin, Gatifloxacin and Moxifloxacin in Serum by Liquid Chromatography with Column Switching,” Journal of Chromatography B. Analytical Technologies in the Biomedical and Life Sciences, Vol. 810, No. 1, 2004, pp. 77-83.
- E. Nemutlu, S. Kir, O. Ozyuncu and M. S. Beksac, “Simultaneous Separation and Determination of Seven Quinolones Using HPLC: Analysis of Levofloxacin and Moxifloxacin in Plasma and Amniotic Fluid,” Chromatographia, Vol. 66, Suppl. 1, 2007, pp. 15-24. doi:10.1365/s10337-007-0292-9
- F. A. Siddiqui, M. S. Arayne, N. Sultana, A. Z. Mirza, F. Qureshi and H. Zuberi, “Facile and Manifest Spectrophotometric Methods for the Determination of Six Quinolone Antibiotics in Pharmaceutical Formulations Using Iron Salts,” Medicinal Chemistry Research, Vol. 19, No. 9, 2009, pp. 1259-1272.
- N. Sultana, N. Shafi, A. Naz and H. Shamshad, “RPHPLC Method for the Simultaneous Determination of Diltiazem and Quinolones in Bulk Formulations and Human Serum,” Journal of Chilian Chemical Society, Vol. 54, No. 4, 2009, pp. 289-293.
- N. Sultana, M. S. Arayne and W. Shehzad, “Simultaneous Determination of Ceftriaxone Sodium and Statins Drugs in Pharmaceutical Formulations and Human Serum by RP-HPLC,” Journal of The Chilean Chemical Society, Vol. 55, No. 2, 2010, pp. 193-198.
- S. Koichi, W. L. Lee, T. Toyohide, S. Yasuhide, S. Kiyohito, T. Yuji and K. Susumu, “Rapid and Simultaneous Determination of Nonsteroidal Anti-Inflammatory Drugs in Human Plasma by LC-MS with Solid-Phase Extraction,” Analytical and Bioanalytical Chemistry, Vol. 384, No. 7-8, 2006, pp. 1501-1505.
- S. Koichi, W. L. Lee, T. Toyohide, S. Yasuhide, S. Kiyohito, T. Yuji and K. Susumu, “On-Line Sample Extraction and Enrichment of Non-Steroidal Anti-Inflammatory Drugs by Pre-Column in Capillary Liquid Chromatography Mass Spectrometry,” Journal of Chromatography B, Vol. 846, No. 1-2, 2007, pp. 176-183. doi:10.1016/j.jchromb.2006.08.041
- K. E. V. Nagoji, S. Vijayasrinivas, M. K. Kumar, N. Mathivanan, M. S. Kumar and M. E. Rao, “Simultaneous Reverse Phase HPLC Estimation Of Nimesulide and Diclofenac Sodium,” Indian Journal of Pharmaceutical Sciences, Vol. 65, No. 4, 2003, pp. 407-409.
- N. Srinivas, L. Narasu, B. P. Shankar and R. Mullangi, “Development and Validation of a HPLC Method for Simultaneous Quantitation of Gatifloxacin, Sparfloxacin and Moxifloxacin Using Levofloxacin as Internal Standard in Human Plasma: Application to a Clinical Pharmacokinetic Study,” Biomedical Chromatography, Vol. 22, No. 11, 2008, pp. 1288-1295. doi:10.1002/bmc.1060
- H. R. N. Marona, J. A. S. Zuanazz and E. E. S. Schapoval “Determination of Sparfloxacin and Its Degradation Products by HPLC,” Journal of Antimicrobial Chemotherapy, Vol. 44, No. 2, 1999, pp. 301-302 doi:10.1093/jac/44.2.301
- A. M. El-Didamony, “Fluorescence Probe Enhanced Spectrofluorimetric Method for the Determination of Sparfloxacin in Tablets and Biological Fluids,” Luminescence, Vol. 26, No. 2, 2010, pp. 112-117. doi:10.1002/bio.1192
- A. M. El-Didamony, “Spectrophotometric Determination Method of Sparfloxacin in Pharmaceutical Preparations by Ternary Complex Formation with Pd(II) and Eosin,” Analytical Letters, Vol. 40, No. 14, 2007, pp. 2708-2720. doi:10.1080/00032710701588408
- H.-Y. Cho, S.-A. Park and Y.-B. Lee, “Improvement and Validation of an HPLC Method for Examining the Effects of the MDR1 Gene Polymorphism on Sparfloxacin Pharmacokinetics,” Journal of Chromatography B, Vol. 834, No. 1-2, 2006, pp. 84-92.
- A. P. Argekar and S. J. Shah, “Stability Indicating HPLC Method for the Determination of Sparfloxacin (Spar),” Analytical Letters, Vol. 32, No. 7, 1999, pp. 1363-1370. doi:10.1080/00032719908542903
- H. R. N. Marona and E. E. S. Schapoval, “A High-Performance Liquid Chromatographic Assay for Sparfloxacin,” Journal of Pharmaceutical and Biomedical Analysis, Vol. 20, No. 3, 1999, pp. 413-417. doi:10.1016/S0731-7085(98)00102-2
- N. N. Rahman and S. Ahmad, “Development of Quantitative Analysis of Sparfloxacin by High Performance Liquid Chromatography,” The Dhaka University Journal of Pharmaceutical Sciences, Vol. 6, No. 1, 2007.
- A. Srikar, K. P. Channabasavaraj, G. Dharmamoorty, N. Valmiki, C. Chinnappa and T .V. Babu, “Spectrophotometric Methods for Quantitative Estimation of Sparfloxacin in Bulk and Pharmaceutical Dosage Forms,” Journal of Pharmaceutical Sciences and Research, Vol. 1, No. 2, 2009, pp. 13-15.
- J. R. Brouwers, “Drug Interactions with Quinolone Antibacterials,” Drug Safely, Vol. 7, No. 4, 1999, pp. 268-281. doi:10.2165/00002018-199207040-00003
- T. Yoshino, M. Noguchi, H. Okutsu, A. Kimoto, M. Sasamata and K. Miyata, “Celecoxib Does Not Induce Convulsions nor Does It Affect GABAA Receptor Binding Activity in the Presence of New Quinolones in Mice,” European Journal of Pharmacology, Vol. 501, No. 1-3, 2005, pp. 69-76. doi:10.1016/j.ejphar.2004.11.038
- F. Leite, “Validation in Chemical Analysis Atom,” 4th Edition, Átomo, São Paulo, 2002, pp. 69-72.
- United States Pharmacopeia, “United States Pharmacopeial Convention,” 21st Edition, Rockville, 2002.
- L. N. Linda and Center for Drug Evaluation and Research (CDER) “Reviewer Guidance: Validation of Chromatographic Methods,” Center for Drug Evaluation and Research, Rockville, 1994.
- M. W. Dong, “Modern HPLC for Practicing Scientists,” John Wiley and Sons, Hoboken, 2006. doi:10.1002/0471973106
- United States Pharmacopeia, 28th Edition, United States Pharmacopeial Convention, Rockville, 2005.
- ICH, “International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use,” Vol. 4, Washington DC, 11-12 September 2002, pp. 69-72.
- Association of Official Analytical Chemists, “Official Methods of Analysis of AOAC International,” 17th Edition, AOAC, Gaithersburg, 2002, p. 20.