Open Journal of Clinical Diagnostics
Vol.09 No.04(2019), Article ID:96582,7 pages
10.4236/ojcd.2019.94010
Congenital Pulmonary Adenomatoid Malformation: Unusual Revelation
M. Echchikhi, H. Essaber, N. Allali, L. Chat
Department of Medical Imaging and Radiodiagnosis, Children’s Hospital, CHU Ibn Sina, Rabat, Morocco
![]()
Copyright © 2019 by author(s) and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY 4.0).
http://creativecommons.org/licenses/by/4.0/



Received: October 10, 2019; Accepted: November 23, 2019; Published: November 26, 2019
ABSTRACT
Congenital pulmonary adenomatoid malformation (CPAM) is a congenital lung malformation that is distinguished by abnormal airway patterning during branching morphogenesis. It may lead to significant morbidity and mortality in infants due to complications such as pulmonary infections, lung hypoplasia, respiratory distress, and fetal hydrops. The diagnosis is usually prenatal thanks to morphological ultrasound; in postnatal, the anomaly may remain asymptomatic or show respiratory signs or complications. The postnatal diagnosis is based on computed tomography. In this article, we report a case of congenital pulmonary adenomatoid malformation in a 7-month-old patient who presented respiratory distress that is due to voluminous infected pleural effusion. The diagnosis of CPAM was suspected at the initial CT and confirmed at the control CT that was realized after treatment of the infection. The therapeutic management of CPAM is depending on the severity of complications and its evolution.
Keywords:
Congenital, Pulmonary, Malformation

1. Introduction
Congenital pulmonary adenomatoid malformation (CPAM) is defined as a congenital disorder of lung parenchyma that results from an abnormal branching of immature lung bronchioles. It can be diagnosed in the fetus or in postnatal period thanks to different imaging means.
It represents 30% to 40% of all congenital lung diseases in foetuses, accounting as the most common diagnosed lung malformation in prenatal [1].
The malformation is also the most common cause of respiratory distress in the neonatal period, and exposes to complications of varying severity that may be life-threatening.
The treatment and the prognosis for CPAM depend on the size of the lesion, the presence of complications or other associated anomalies.
We report a case of unusual manifestation of CPAM in a 7-month infant, and highlight the role of computed tomography for postnatal diagnosis of this malformation.
2. Observation
A 7-month male infant with not tracked pregnancy history and no known history of any disease, he was brought to the emergency for acute dyspnea.
Clinical examination found respiratory distress, generalized hypotonia, and fever at 39˚C.
Resuscitative measures were taken and explorations were launched. Blood laboratory tests showed leukocytosis at 20,000/ml and elevated C-Reactive Protein at 93 mg/L.
The thoracic X-Ray showed opaque right hemithorax, CT scan was required for better characterization, which objectified an abundant right pleural effusion with air-fluid level and millimetric air bubbles coming out of the collapsed lung, suspecting a broken congenital pulmonary airway malformation (Figure 1); the

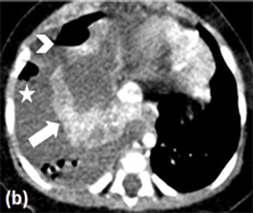

Figure 1. Thoracic contrast CT in axial (a, b) and coronal (c) sections, showing an important right pleural effusion (asterisk) with air-fluid level (arrow head); the right lung parenchyma is collapsed (thick arrow) with adjacent air bubbles (thin arrow), raising the hypothesis of a broken congenital pulmonary airway malformation.
first malformations evoked were congenital pulmonary adenomatoid malformation and bronchopulmonary sequestration.
Pleural fluid was drained twice with a positive bacteriological examination; the infant was treated with antibiotics that led to satisfactory clinical and biological evolution.
Control CT scan (Figure 2) showed the disappearance of pleural effusion and demonstrated a right lung lesion which measuring 21 × 15 × 18 mm, and is made of contiguous microcysts with liquid and aerial content, without other notable abnormality especially no systemic arterial branch; that confirmed the diagnosis of congenital pulmonary adenomatoid cystic malformation.
The therapeutic decision taken after a multidisciplinary staff consisted of a therapeutic abstention and a close follow-up of the infant, since the small size of the lesion, the favorable evolution of symptoms, and the absence of recidivism.
3. Discussion
Congenital pulmonary adenomatoid malformation (CPAM) is a rare condition




Figure 2. Control thoracic CT without contrast done 1 month after the end of the antibiotic treatment. (a) and (b): mediastinal window in axial (a) and coronal (b) sections, showing the total disappearance of pleural effusion. (c) and (d): images using lung window settings demonstrates a lesion in the mediobasal segment of the right lung, which is made of contiguous microcysts with liquid and aerial content, the lesion measures 21 × 15 × 18 mm. Absence of systemic arterial branch or other notable abnormality. The lesion found is compatible with congenital pulmonary adenomatoid cystic malformation.
with a documented incidence ranging from 1 in 11,000 to 35,000 live births, with a male predominance. 85% to 95% of cases are unilobar, it is defined as a heterogeneous group of cystic and non-cystic lung lesions resulting from early airway maldevelopment [2].
Congenital pulmonary airway malformations previously called congenital cystic adenomatoid malformations (CCAM), this term was inaccurate and needed to be changed because not all types of malformations are cystic, only one is adenomatoid and all of them have anatomic and microscopic features of pulmonary airways.
Stocker proposed the new name in 2002 and expanded his three types classification to five types: type 0 consists of acinar dysgenesis or dysplasia, type 1 represents large cysts, type 2 corresponds to numerous small cysts less than 1 mm, type 3 represents microcystic lesions, and type 4 consists of distal acinar origin [3] [4].
The malformation is formed by abnormal branching of immature bronchioles, microscopic features of CPAM include polypoid projections of the mucosa, presence of inflammatory cells, hypertrophy of smooth muscle and elastin within cyst walls, absence of cartilage and the presence of mucous-secreting cells. It derives their vascularisation from normal pulmonary circulation [4].
Genes thought to contribute to the pathogenesis of CPAM includes HOXB5 and platelet-derived growth factor-B (PDGFB). These genes are of clinical importance as they may lead to the development of future therapeutic strategies [5] [6].
CPAM can have various symptoms ranging from asymptomatic course to dyspnea, cyanosis and respiratory distress in neonates and children. Asymptomatic malformations may remain undetected until adulthood when finding accidentally or occur as recurrent pulmonary infections.
80% of cases reported in literature presents with respiratory symptoms at birth or during their first month of life, the most common symptom in newborns is respiratory distress related to lung hypoplasia [4] [7] [8].
Later presentations of CPAM in infants are rare, and include recurrent pneumonia, pneumothorax, lung abscess, and reactive airway disease [9].
A broken malformation leading to abundant and infected pleural effusion as reported in this article is an unusual revelation of CPAM.
Associated anomalies have been reported in literature including heart, neural tube, renal, facial and pulmonary malformations, the most frequently found is the mixed CPAM-bronchopulmonary sequestration “hybrid lesion” [2] [9].
CPAM might be diagnosed during prenatal ultrasound (PUS) as early as 12 weeks of gestation but are usually found at a scheduled prenatal check-up at 18 - 23 weeks [2] [10].
The typical finding is a solid hyperechoic or cystic lung mass within the thoracic cavity. Ultrasound in the second trimester accurately detects echogenic pulmonary lesions, assesses lung size and degree of mediastinal shift as well as the presence of polyhydramnios and hydrops, it may detect a systemic arterial supply on Doppler which is a component of bronchopulmonary sequestration in mixed CPAM-sequestration “hybrid lesion”.
Prenatal MRI has been shown to be highly accurate in defining congenital lung anomalies (T2SSFSE is the most useful sequence); solid and/or microcystic lesions almost higher in signal intensity than remaining lungs. It evaluates the hypoplasia of remaining lungs, mass effect on mediastinal structures, presence of additional anomalies and finds an associated hydrops [10] [11].
Neonatal chest radiography is usually the first investigation method performed, but its sensitivity is also not very high, especially in asymptomatic patients. CPAM appears as a hyper lucent region or multiple various lesions air-filled with thin-walled.
CT has higher sensitivity and specificity than prenatal ultrasound and neonatal chest radiography which makes CT the method of choice to diagnose congenital pulmonary malformations in newborn and child.
It is indicated when diagnostic uncertainty on plain radiograph, symptomatic children, and pre-surgical planning. It can evaluate the extent of the lesion, precisely describe cyst size and wall thickness, the type of blood supply (systemic or pulmonary), which not only helps to differentiate CPAM from other lesions but also is essential prior to surgical intervention. CT is also used to diagnose complications such as pneumothorax (especially if it is small and not visible on radiography), and infections [10] [11].
Several differential diagnoses can be discussed:
· Bronchopulmonary sequestration: it affects more left lower lobes with solid-appearing; it only becomes air-filled after infection. Mixed CPAM/sequestration or “hybrid lesions” have more likely discrete cysts.
· Congenital diaphragmatic hernia: appears radiographically as multicystic and air-containing mass, cysts often uniform in size and morphology; we find also a paucity of abdominal bowel gas (due to herniation).
· Bronchogenic cyst: is often round, typically mediastinal or perihilar, as a fluid-density mass.
· Pleuropulmonary blastoma: very rare neoplasm of young children; typically not discovered in prenatal or neonatal periods.
· Congenital lobar overinflation: is progressive lobar overexpansion in fetus or neonate, rarely occurs in lower lobes.
· Persistent pulmonary interstitial emphysema: may appear as a focal cyst or regional small bubbly lesions [10] [12].
The therapeutic management of CPAM can be pharmacologic (treatment of complications like our case), surgical or simple follow-up, depending on the severity of the disease and its evolution. Surgical resection is required if CPAM is symptomatic without improvement. The asymptomatic form has controversial managements according to different teams, most advocate elective resection particularly with the notion of pulmonary compensation in children that avoid risks of infection and malignant evolution [12] [13] [14].
In our patient, the favorable evolution of symptoms, the absence of recidivism, and the small size of the lesion have oriented more toward a close follow-up of the child.
4. Conclusions
Congenital pulmonary adenomatoid malformations are detected more frequently in the era of prenatal ultrasound.
The postnatal revelation of the malformation is variable; it is rarely as an infected pleural effusion, as the case of our patient. The postnatal diagnosis is based on computed tomography that also confirms the malformation and eliminates differential diagnoses.
Currently, there is no unified strategy for treatment, then, the management decisions vary among institutions.
Conflicts of Interest
The authors declare no conflicts of interest regarding the publication of this paper.
Cite this paper
Echchikhi, M., Essaber, H., Allali, N. and Chat, L. (2019) Congenital Pulmonary Adenomatoid Malformation: Unusual Revelation. Open Journal of Clinical Diagnostics, 9, 145-151. https://doi.org/10.4236/ojcd.2019.94010
References
- 1. Hansell, D.M., Bankier, A.A., MacMahon, H., et al. (2008) Fleischner Society: Glossary of Terms for Thoracic Imaging. Radiology, 246, 697-722. https://doi.org/10.1148/radiol.2462070712
- 2. Azizkhan, R.G. and Crombleholme, T.M. (2008) Congenital Cystic Lung Disease: Contemporary Antenatal and Postnatal Management. Pediatric Surgery International, 24, 643-657. https://doi.org/10.1007/s00383-008-2139-3
- 3. Stocker, J.T. (2002) Congenital Pulmonary Airway Malformation—A New Name for and an Expanded Classification of Congenital Cystic Adenomatoid Malformation of the Lung. Histopathology, 41, 424-430.
- 4. Stocker, J.T., Madewell, J.E. and Drake, R.M. (1977) Congenital Cystic Adenomatoid Malformation of the Lung: Classification and Morphologic Spectrum. Human Pathology, 8, 155-171. https://doi.org/10.1016/S0046-8177(77)80078-6
- 5. Volpe, M.V., Pham, L., Lessin, M., Ralston, S.J., et al. (2003) Expression of Hoxb-5 during Human Lung Development and in Congenital Lung Malformations. Birth Defects Research Part A: Clinical and Molecular Teratology, 67, 550-556. https://doi.org/10.1002/bdra.10086
- 6. Liechty, K.W., Crombleholme, T., Quinn, T.M., Cass, D.L., et al. (1999) Elevated Platelet-Derived Growth Factor-B in Congenital Cystic Adenomatoid Malformations Requiring Fetal Resection. Journal of Pediatric Surgery, 34, 805-809. https://doi.org/10.1016/S0022-3468(99)90377-9
- 7. Cloutier, M.M., Schaeffer, D.A. and Hight, D. (1993) Congenital Cystic Adenomatoid Malformation. Chest, 103, 761-764. https://doi.org/10.1378/chest.103.3.761
- 8. Bianchi, D.W., Crombleholme, T.M. and D’Alton, M.E. (2000) Cystic Adenomatoid Malformation. In: Fetology: Diagnosis and Management of the Fetal Patient, Chapter 37, McGraw-Hill, New York.
- 9. Pinkerton, H.J. and Oldham, K.T. (2005) Principles and Practice of Pediatric Surgery. Chapter 61, Volume 2, Springer, Berlin, Heidelberg.
- 10. Pacharn, P., Kline-Fath, B., Calvo-Garcia, M., Linam, L.E., Rubio, E.I., Salisbury, S. and Brody, A.S. (2013) Congenital Lung Lesions: Prenatal MRI and Postnatal Findings. Pediatric Radiology, 43, 1136-1143. https://doi.org/10.1007/s00247-013-2668-3
- 11. An, P., Wang, Y., Feng, W., et al. (2019) Congenital Cystic Adenomatoid Malformation Volume Ratio in Prenatal Assessment of Prognosis of Fetal Pulmonary Sequestrations. Current Medical Science, 39, 658-662. https://doi.org/10.1007/s11596-019-2088-1
- 12. Annunziata, F., Bush, A., Borgia, F., et al. (2019) Congenital Lung Malformations: Unresolved Issues and Unanswered Questions. Frontiers in Pediatrics, 7, 239. https://doi.org/10.3389/fped.2019.00239
- 13. Kapralik, J., Wayne, C., Chan, E. and Nasr, A. (2016) Surgical versus Conservative Management of Congenital Pulmonary Airway Malformation in Children: A Systematic Review and Meta-Analysis. Journal of Pediatric Surgery, 51, 508-512. https://doi.org/10.1016/j.jpedsurg.2015.11.022
- 14. Frick, A.E., Decaluwe, H., Weynand, B., et al. (2019) Invasive Mucinous Adenocarcinoma of the Lung Arising in a Type1 Congenital Pulmonary Airway Malformation in a 68-Year-Old Patient: A Case Report. Acta Chirurgica Belgica, 2, 1-6.