Journal of Cancer Therapy
Vol.2 No.2(2011), Article ID:5809,8 pages DOI:10.4236/jct.2011.22033
Tumor Resistance to Molecularly Targeted Agents
![]()
1Light Sciences Oncology, Inc., Bellevue, USA;
2Florida Hospital Waterman, Tavares, USA;
3Division of Cancer Studies, University of Birmingham, Birmingham, UK;
4Department of Radiation Oncology, Abramson Cancer Center of the University of Pennsylvania, Philadelphia, USA;
5Division of Surgery, Singapore General Hospital, Singapore City, Singapore.
Email: jamesc@lsoncology.com
Received October 10th, 2011; revised March 9th, 2011; accepted March 20th, 2011.
Keywords: Tumor Resistance, Signaling Pathway, Gene Pathway, Oncogene, Renal Cell Carcinoma
ABSTRACT
Despite the development of new targeted cancer therapies, primary and secondary tumors continue to be a leading cause of suffering and mortality worldwide. The complexity and heterogeneity of malignancies ultimately result in tumor resistance to therapies, such as EGFR inhibitiors, anti-angiogenesis agents, and tyrosine kinase inhibitors. An example of targeted therapies for renal cell carcinoma is presented, including the SAE profile and efficacy of 6 targeted agents. Although survival is improved, resistance and an increased side effect profile diminish the benefits of these agents. New agents are needed which are pathway independent and which can overcome tumor resistance without adding to the side effect profile of current therapies.
1. Introduction
Uncontrolled primary and metastatic cancer continues to take a serious toll on Americans, with one in four deaths in the United States due to malignancy (562,340 deaths predicted in 2009) [1,2], and over 4 million people under care for cancer and its sequelae [3]. This significant cancer death rate, given the panoply of expensive molecularly targeted drugs designed to inhibit various cancer growth and development pathways, attests to the complexity and heterogeneity of malignant disease [4-8] . Established cancers rapidly develop resistance to the blockade of a single pathway or a limited set of pathways, and any singular targeting approach is unlikely to result in dramatic improvements for the majority of patients with advanced disease [9,10]. Even when different combinations of systemic agents are used against solid tumors, the survival gains may be minimal and are often associated with increased side effects and significant financial burden [11,12] .
The elucidation of the genetic diversity of different cancers has led to the creation of gene and protein databases together with maps of signaling proteins and pathways of increasing complexity [13,14] . The routine discovery of new signaling proteins and pathways has allowed for the continuous development of targeted chemotherapeutic agents designed to block the communication activity within specific pathways. However, genetic instability inherent within cancer cells produces innate and acquired resistance to systemic therapies, including molecularly targeted agents. Enormous effort and expenditure has resulted in some meaningful therapeutic advances, but for the majority of solid tumor patients with metastatic disease, the outlook remains grim.
2. Complex Tumor Progression Pathways
An obstacle to the current approach of blocking a limited set of essentially linear pathways is the evolution of highly sophisticated communication networks comprised of a myriad of proteins encoded by aberrant and dysfunctionally expressed genes. The detailed human oncogene-signaling map generated by Cui et al. (Figure 1) exemplifies the complexity of the architecture and relationships between large numbers of proteins involved in tumor survival and progression [13].
3. The Connections of a Single Protein
The complex interaction network of a single protein involved in metastasis, HEF 1 (Human Enhancer of Filimentation), further illustrates the redundancy and intricate connectivity typical of tumor survival mechanisms. The web of connections of this protein which is involved in cell attachment, motility, and oncogenic transformation can be viewed in Cytoscope [15].

Figure 1. Human Oncogene-Signaling Map [13]. The human oncogene-signaling map was extracted from the human signaling network, which was mapped with mutated and methylated cancer genes. The map displays three “oncogenic-dependent regions” (three light gray regions in background) in which genes of the two regions are also heavily methylated. Nodes represent genes, whereas the links with and without arrows represent signal and physical relations, respectively. Nodes in red, purple, brown, cyan, blue and green represent the genes that are highly mutated but not methylated, both highly mutated and methylated, poorly mutated but not methylated, both poorly mutated and methylated, methylated but not mutated, and neither mutated nor methylated, respectively. Reprinted by permission from Macmillan Publishers Ltd: Molecular Systems Biology, Copyright 2007 [13].
4. Resistance Mechanisms to EGFR Inhibitors
The multiple links and interconnectivity of signaling proteins confers numerous parallel pathways and alternative routes by which tumors may maintain activities which further malignant development and spread in the face of modern combination therapy. An analysis of how networks of proteins confer resistance to epidermal growth factor receptor (EGFR) inhibitors has recently been published [16].
5. Resistance to Anti-Angiogenesis Drugs
Resistance mechanisms to anti-angiogenesis drugs, such as the VEGF neutralizing antibody bevacizumab, have also been elucidated. Intrinsic non-responsiveness is present, alternative pathway activation occurs, and solid tumors may then adopt a more invasive pattern of tumor growth [17,18]. Increased invasiveness occurs due to altered expression of angiogenesis and invasion-related genes [18]. Pro-angiogenic and immune suppressive bFGF and many other pro-angiogenic signaling proteins are upregulated after chronic therapy with bevacizumab-type inhibitors, leading to increased intratumoral microvessel density in tumor models [17,18]. Tumor invasiveness is enhanced by upregulation of various matrix metallopeptidases [18]. Physical changes in tumor vasculature, including pericyte coverage and recruitment of bone marrow-derived pro-angiogenic cells, occur, which obviate the need for VEGF mediated angiogenesis and vascular survival, leading to relapse [17].
Ebos et al. have further classified resistance mechanisms in terms of those generated by the tumor itself and those mediated by the host [19].
6. Resistance to Tyrosine Kinase Inhibitors
Similarly to anti-angiogenic agents, tyrosine kinase inhibitors such as erlotinib and gefitinib have been introduced into the clinical setting, demonstrating modest results for most patients and an association with agent resistance. [20] Many tumors are innately resistant, and multiple mechanisms of acquired resistance occur: 1) secondary EGFR mutations; 2) amplification of MET, a tyrosine kinase receptor; 3) loss of IGF-binding proteins; and 4) activation of other alternative downstream signaling pathways and pathway switching [20-23]. Selective pressure occurs for cells to acquire resistance through mutations in the kinase gene that reduce or prevent inhibitor drug binding [24].
Inherent genetic instability within cancers also confers an added temporal dimension contributing to cell diversity. Even single tumors are typically heterogeneous and shifting in pathway activation states over time [25,26].
Malignant tumors may be intrinsically resistant to molecularly targeted agents or may rapidly acquire resistance via a variety of mechanisms, even when combination therapy is used. Survival improvement from signal blockade in patients with metastatic disease has been modest due in large part to the sheer number and redundancy of alternate tumor survival and growth pathways. New approaches that globally block protein signaling and other related tumor promoting processes need to be developed. However, such global approaches cannot be applied systemically, since overall cell growth would be inhibited, and thus such blocking drugs would not be compatible with life or would produce intolerable side effects.
7. Renal Cell Carcinoma as an Example of Resistance to Six Targeted Drugs
Renal cell carcinoma (RCC) is the seventh most common cancer in men and the ninth most common cancer in women, with over 209,000 newly diagnosed cases and over 102,000 deaths annually worldwide [27]. Though some RCCs are secondary to heritable genetic changes, the majority of RCC patients present with sporadic (non-inherited) tumors secondary to acquired defects in both VHL alleles, resulting in an abnormal von Hippel-Lindau protein. The altered protein leads to development of a vascular tumor with significant metastatic potential [27]. RCC is highly resistant to both chemotherapy and radiotherapy. Surgery may be curative in some patients, but many recur, and RCC is often locally advanced, metastatic, or even unresectable at the time of diagnosis. A small minority of patients develop a spontaneous immune response, the effect of which may be dramatic. To increase this favorable response, immunotherapy (mainly high dose bolus interleulin-2 [IL-2]) has been developed and approved by the U.S. Food and Drug Administration based on the potential for durable complete remission in approximately 10% of treated patients. High dose IL-2, however, is associated with severe toxicity and requires hospitalization and specialized care during administration [28] Recently, the therapy for nonoperable metastatic RCC has undergone a rapid evolution, and chemotherapeutics, radiotherapy, and toxic immunotherapy have largely yielded to a generation of molecularly targeted agents (listed in Table 1).
The pathway inhibitors listed in Table 1 inhibit tumor angiogenesis by blocking signaling pathways. As a whole, the action of blocking specific growth and development pathways is not confined to the malignancy, but also affects normal cells. Thus, a myriad of side effects, some of which overlap with those associated with chemotherapy and some of which are new, are associated with administration of these drugs (Table 3).
A common current recommendation for initial therapy of systemic RCC is sunitinib monotherapy for “better risk” patients and temsirolimus for “poor risk” patients [29,30] Sorafenib is advised as second-line therapy [31]. Bevacizumab, interferon (IFN), and newly approved everolimus are also options [32] . Response rates and survival are summarized in Table 2.
The introduction of targeted agents has improved overall survival (OS) significantly when compared to older immunotherapy and chemotherapy. However, associated toxicity, including a significant incidence of severe adverse events, remains a concern with utilization of these drugs and has been shown to be additive in

Table 1. Targeted agents utilized in metastatic renal cell carcinoma.

Table 2. Activity of Targeted Agents [31,33-35].

Table 3. Incidence of Serious Adverse Events [28,32,33,35].
combination therapy trials [32,36] .
Resistance to targeted agents clearly occurs with all mono and combined therapy, as well as with sequenced therapy.
8. Combination Therapy
Side effects are very common with targeted agents, and studies employing combination therapy aimed at overcoming pathway redundancy and resistance have sometimes proven problematic. For example, the combination of suntinib and bevacizumab has been assessed in a phase 1 study; unfortunately, grade 3 - 4 side effects were very common, and the combination proved to be too toxic [33]. Similarly, sunitinib and temsirolimus have been studied in combination, and the trial was terminated prematurely after two of the first three enrolled patients required hospitalization for severe toxicity [33].
The combination of bevacizumb and everolimus has been evaluated in a phase 2 study. While overall RR was 21%, grade 3 - 4 side effects occurred in 19% of patients (proteinuria 19%, fatigue 9%, stomatitis 8%) [33]. Median PFS was 9 months in naïve and 6 months in pretreated patients [33]. A number of other bevacizumab combinations have necessitated dose reductions in response to enhanced toxicities of the second agent, negating the potential beneficial effects of the combined therapy [37].
9. Resistance to mTor Inhibition
The mTor related signaling and growth pathways involved with solid tumor development and progression are exceedingly complicated and are depicted in other maps [38]. Review of the KEGG (Kyoto Encyclopedia of Genes and Genomes) cancer pathway map example (Figure 2) illustrates that inhibition of mTOR potentially leaves 14 other redundant pathways intact, all preventing tumor cell apoptosis. Similarly, angiogenesis is sustained by pathways other than those blocked by kinase and VEGF inhibitors. Combination therapy intended to block more signaling pathways has proven in some studies to be toxic or improves outcomes marginally if at all relative to monotherapy. Treatment resistance typically develops within 12 months or less, and tumors tend to progress rapidly after therapy is terminated [39] . Exposing patients to the toxicity of two combined drugs versus sequential drug treatment in an attempt to overcome resistance is unlikely to be clinical practical [40].
Depicted in Figure 3 are predicted resistance points to rapamycin analogues temsirolimus and everolimus [42]. Table 4 describes the mechanisms of mTOR inhibition resistance.
Prior to the introduction of targeted agents, median OS was only 10 to 13 months after progression on cytokine based therapy [44]. Patients with good prognostic factors who are younger and able to tolerate targeted agents may achieve almost a doubling of median OS. With six targeted pathway inhibitors in use, current strategies may be reaching the limit in terms of patient benefit. Compared to the older immunotherapies, which did produce complete disease clearance in a minority of patients (albeit with extreme toxicity), complete response is virtually never achieved with molecularly targeted agents [33,45] . Trials of combination therapy using newer targeted agents have frequently been problematic due to additive toxicities, and thus sequencing of agents is recommended [40,46].
Is a plateau being reached regarding the clinical benefit that can be expected from development of new targeted agents that follow the paradigm of limited pathway blockade? Elucidation of the multiplicity of intercomnecting cancer growth and development pathways has aided understanding of the mechanisms behind compensatory tumor survival responses. The inhibition of VEGF and mTOR pathways triggers gene expression and upregulation of other proteins that enable tumor cellular and angiogenenic escape [27]. Development of further related agents may not lead to major improvements in survival (Table 4).
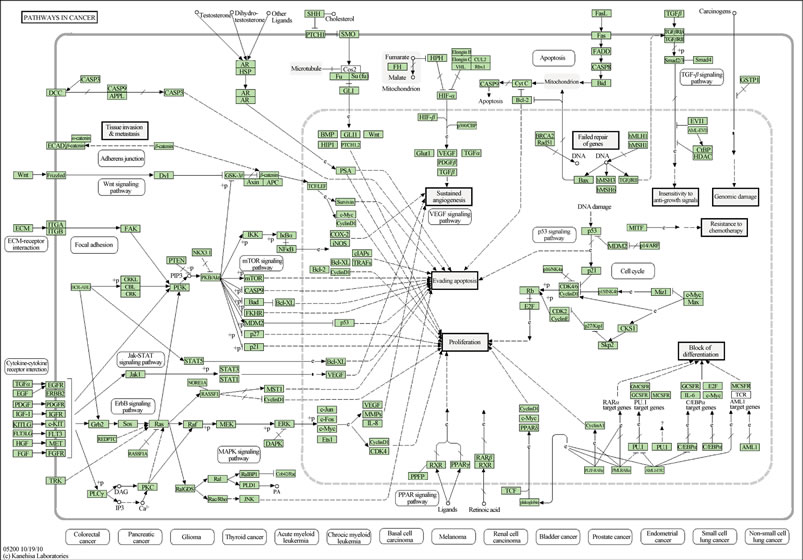
Figure 2. KEGG (Kyoto Encyclopedia of Genes and Genomes) cancer pathway map. Reprinted by permission from GenomeNet [41].

Figure 3. (a). Predicted Mechanisms of Resistance to Rapamycin Analogues. Rapamaycin or its derivatives (red balls) can be eliminated from cells by ABC transporters such as P-glycoprotein. Mutations of FKBP or mTOR (yellow stars) confer resistance. Acquired resistance to rapamycin has been associated with decreased stoichiometry between 4E-BP and eIF4E, either through decreased translation of 4E-BP or via overexpression of eIF4E. (b). Inhibition of mTOR leads to decreased translation of cyclin D1 mRNA and reduced levels of cyclin D1. In many cells, there is a concomitant stabilization of the cyclin-dependent kinase inhibitor p27Kip1 inhibition of CDK-cyclin activity, and decreased phosphorylation of RB. Cells deficient in p27Kip1 are partially resistant to inhibition of proliferation by rapamycin, whereas RB-null cells are completely resistant. Reprinted by permission from Macmillan Publishers Ltd: Br J Cancer, copyright 2006 [42].

Table 4. Mechanisms of mTOR intrinsic or acquired resistance.
10. Conclusions
The advent of the molecularly targeted drug era has led to meaningful increases in survival for some specific cancer patients enrolled in clinical trials. However, many patients with advanced cancers do not fit the relatively narrow profile of enrolled clinical trial patients and experience worse outcomes when compared to published study results [49]. In addition, a higher incidence of serious toxicities associated with these agents has been demonstrated as larger numbers of patients are treated in post approval studies [50,51]. Resistance will develop in the vast majority of patients with metastatic disease, and disease progression is still the rule. Furthermore, with new agents also come new classes of side effects, and some patients will not tolerate another sequential agent at the time of progression or the demonstration of intolerance to the first agent. Newer targeted agents that block the same or different pathways as other existing drugs are not likely to represent major advances in survival given the complexity and redundancy of the cancer pathway network. Truly novel treatments which accurately and globally block tumor development and growth pathways with minimal associated toxicity require additional focus and consideration, as this description represents the best hope for many cancer patients.
REFERENCES
- E. Finlay and D. Casarett, “Making Difficult Discussions Easier: Using Prognosis to Facilitate Transitions to Hospice,” CA: A Cancer Journal for Clinicians, Vol. 59, No. 4, 2009, pp. 250-263. doi:10.3322/caac.20022
- A. Jemal, R. Siegel, E. Ward, Y. Hao, J. Xu and M. J. Thun, “Cancer statistics, 2009,” CA: A Cancer Journal for Clinicians, Vol. 59, No. 4, 2009, pp. 225-249. doi:10.3322/caac.20006
- M. J. Horner, L. A. G. Ries, M. Krapcho, N. Neyman, R. Aminou, N. Howlader, S. F. Altekruse, E. J. Feuer, L. Huang, A. Mariotto, B. A. Miller, D. R. Lewis, M. P. Eisner, D. G. Stinchcomb and B. K. Edwards, “SEER Cancer Statistics Review, 1975-2006,” National Cancer Institute, 2009. http://seer.cancer.gov/csr/1975_2006/
- S. Begley, “We fought cancer...and cancer won,” 2008. http://www.newsweek.com/id/157548
- F. Grizzi and M. Chiriva-Internati, “Cancer: looking for simplicity and finding complexity,” Cancer Cell International, Vol. 6, 2006, p. 4. doi:10.1186/1475-2867-6-4
- B. T. Hennessy, A. M. Gonzalez-Angulo, M. S. Carey and G. B. Mills, “A Systems Approach to Analysis of Molecular Complexity in Breast Cancer,” Clinical Cancer Research, Vol. 15, No. 2, 2009, pp. 417-419. doi:10.1158/1078-0432.CCR-08-2620
- G. Kolata, “As Other Death Rates Fall, Cancer’s Scarcely Moves,” New York Times, 24 April 2009, p. A17.
- A. E. Teschendorff, M. Journee, P. A. Absil, R. Sepulchre and C. Caldas, “Elucidating the Altered Transcriptional Programs in Breast Cancer Using Independent Component Analysis,” PLoS Computational Biology, Vol. 3, No. 8, 2007, p. e161. doi:10.1371/journal.pcbi.0030161
- M. Bredel, D. M. Scholtens, G. R. Harsh, C. Bredel, J. P. Chandler, J. J. Renfrow, A. K. Yadav, H. Vogel, A. C. Scheck, R. Tibshirani and B. I. Sikic, “A Network Model of a Cooperative Genetic Landscape in Brain Tumors,” The Journal of the American Medical Association, Vol. 302, No. 3, 2009, pp. 261-275. doi:10.1001/jama.2009.997
- G. Tonon, “From Oncogene to Network Addiction: The New Frontier of Cancer Genomics and Therapeutics,” Future Oncology, Vol. 4, No. 4, 2008, pp. 569-577. doi:10.2217/14796694.4.4.569
- L. B. Saltz, “Progress in Cancer Care: The Hope, the Hype, and the Gap between Reality and Perception,” Journal of Clinical Oncology, Vol. 26, No. 31, 2008, pp. 5020-5021. doi:10.1200/JCO.2008.17.6198
- D. J. Stewart and R. Kurzrock, “Cancer: The Road to Amiens,” Journal of Clinical Oncology, Vol. 27, No. 3, 2009, pp. 328-333. doi:10.1200/JCO.2008.18.9621
- Q. Cui, Y. Ma, M. Jaramillo, H. Bari, A. Awan, S. Yang, S. Zhang, L. Liu, M. Lu, M. O’Connor-McCourt, E. O. Purisima and E. Wang, “A Map of Human Cancer Signaling,” Molecular Systems Biology, Vol. 3, 2007, p. 152. doi:10.1038/msb4100200
- Y. J. Huang, D. Hang, L. J. Lu, L. Tong, M. B. Gerstein and G. T. Montelione, “Targeting the Human Cancer PathWay Protein Interaction Network by Structural Genomics,” Molecular and Cellular Proteomics, Vol. 7, No. 10, 2008, pp. 2048-2060. doi:10.1074/mcp.M700550-MCP200
- E. A. Golemis, I. G. Serebriiskii, N. Skobeleva, E. N. Pugacheva, M. K. Singh, N. Tikhmyanova, E. Izumchenko, S. Bluestein, W. Gao and W. Yun, “The Role of Signaling Hubs in Metastasis,” 2006. www.fccc.edu/docs/sci_report/Golemis.pdf
- E. A. Hopper-Borge, R. E. Nasto, V. Ratushny, L. M. Weiner, E. A. Golemis and I. Astsaturov, “Mechanisms of Tumor Resistance to EGFR-Targeted Therapies,” Expert Opinion on Therapeutic Targets, Vol. 13, No. 3, 2009, pp. 339-362. doi:10.1517/14712590902735795
- G. Bergers and D. Hanahan, “Modes of Resistance to Anti-Angiogenic Therapy,” Nature Reviews Cancer, Vol. 8, No. 8, 2008, pp. 592-603. doi:10.1038/nrc2442
- A. K. Lucio-Eterovic, Y. Piao and J. F. de Groot, “Mediators of Glioblastoma Resistance and Invasion during Antivascular Endothelial Growth Factor Therapy,” Clinical Cancer Research, Vol. 15, No. 14, 2009, pp. 4589-4599. doi:10.1158/1078-0432.CCR-09-0575
- J. M. Ebos, C. R. Lee and R. S. Kerbel, “Tumor and Host-Mediated Pathways of Resistance and Disease Progression in Response to Antiangiogenic Therapy,” Clinical Cancer Research, Vol. 15, No. 16, 2009, pp. 5020-5025. doi:10.1158/1078-0432.CCR-09-0095
- J. A. Engelman and P. A. Janne, “Mechanisms of Acquired Resistance to Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors in Non-Small Cell Lung Cancer,” Clinical Cancer Research, Vol. 14, No. 10, 2008, pp. 2895-2899. doi:10.1158/1078-0432.CCR-07-2248
- M. Guix, A. C. Faber, S. E. Wang, M. G. Olivares, Y. Song, S. Qu, C. Rinehart, B. Seidel, D. Yee, C. L. Arteaga and J. A. Engelman, “Acquired Resistance to EGFR Tyrosine Kinase Inhibitors in Cancer Cells is Mediated by Loss of IGF-Binding Proteins,” The Journal Clinical Investigation, Vol. 118, No. 7, 2008, pp. 2609- 2619.
- H. E. Jones, J. M. Gee, I. R. Hutcheson, J. M. Knowlden, D. Barrow and R. I. Nicholson, “Growth Factor Receptor Interplay and Resistance in Cancer,” Endocrine-Related Cancer, Vol. 13, 2006, pp. S45-51. doi:10.1677/erc.1.01275
- A. Maleddu, M. A. Pantaleo, M. Nannini, M. di Battista, M. Saponara, C. Lolli and G. Biasco, “Mechanisms of Secondary Resistance to Tyrosine Kinase Inhibitors in Gastrointestinal Stromal Tumours (Review),” Oncology Reports, Vol. 21, No. 6, 2009, pp. 1359-1366. doi:10.3892/or_00000361
- J. Zhang, P. L. Yang and N. S. Gray, “Targeting Cancer with Small Molecule Kinase Inhibitors,” Nature Reviews Cancer, Vol. 9, No. 1, 2009, pp. 28-39. doi:10.1038/nrc2559
- A. P. Crijns, R. S. Fehrmann, S. de Jong, F. Gerbens, G. J. Meersma, H. G. Klip, H. Hollema, R. M. Hofstra, G. J. te Meerman, E. G. de Vries and A. G. van der Zee, “Survival-Related Profile, Pathways, and Transcription Factors in Ovarian Cancer,” PLoS Medicine, Vol. 6, No. 2, 2009, p. e24. doi:10.1371/journal.pmed.1000024
- S. Klein and A. Levitzki, “Signal Transduction Therapy for Cancer—Whither Now?” Current Signal Transduction Therapy, Vol. 1, No. 1, 2006, pp. 1-12. doi:10.2174/157436206775269244
- B. I. Rini, S. C. Campbell and B. Escudier, “Renal cell carcinoma,” The Lancet, Vol. 373, No. 9669, 2009, pp. 1119-1132. doi:10.1016/S0140-6736(09)60229-4
- M. B. Atkins, “Molecularly Targeted Therapy for Advanced Renal Cell Carcinoma,” 2009. http://www.utdol.com/home/content/topic.do?topicKey=gucancer/14941&view=print
- T. K. Choueiri, “Metastatic Renal Cell Carcinoma: A Guide to Therapy Based on Current Evidence,” Urology Annals, Vol. 1, No. 1, 2009, pp. 9-14. doi:10.4103/0974-7796.48781
- Canadian Kidney Cancer Forum 2009, “Management of kidney cancer: Canadian Kidney Cancer Forum Consensus Update,” Canadian Urological Association Journal, Vol. 3, No. 3, 2009, pp. 200-204.
- J. Bellmunt and M. Guix, “The Medical Management of Metastatic Renal Cell Carcinoma: Integrating New Guidelines and Recommendations,” British Journal of Urology International, Vol. 103, No. 5, 2009, pp. 572-577. doi:10.1111/j.1464-410X.2008.08336.x
- C. Porta and C. Szczylik, “Tolerability of First-Line Therapy for Metastatic Renal Cell Carcinoma,” Cancer Treat Reviews, Vol. 35, No. 3, 2009, pp. 297-307. doi:10.1016/j.ctrv.2008.12.003
- M. Basso, A. Cassano and C. Barone, “A Survey of Therapy for Advanced Renal Cell Carcinoma,” Urologic Oncology, Vol. 28, No. 2, 2009, pp. 121-133. doi:10.1016/j.urolonc.2009.04.015
- National Cancer Institute, “FDA Approval for Everolimus,” 2009. http://www.cancer.gov/cancertopics/druginfo/fda-everolimus
- GlaxoSmithKline, “FDA Approves GlaxoSmithKline’s Votrient™ for Advanced Renal Cell Cancer,” 2009. http://www.gsk.com/media/pressreleases/2009/2009_pressrelease_10113.htm
- P. H. Patel, P. L. Senico, R. E. Curiel and R. J. Motzer, “Phase I Study Combining Treatment with Temsirolimus and Sunitinib Malate in Patients with Advanced Renal Cell Carcinoma,” Clinical Genitourinary Cancer, Vol. 7, No. 1, 2009, pp. 24-27. doi:10.3816/CGC.2009.n.004
- G. Di Lorenzo, R. Autorino and C. N. Sternberg, “Metastatic Renal Cell Carcinoma: Recent Advances in the Targeted Therapy Era,” European Urology, Vol. 56, No. 6, 2009, pp. 891-1104.
- J. B. Easton, R. T. Kurmasheva and P. J. Houghton, “IRS-1: Auditing the Effectiveness of mTOR Inhibitors,” Cancer Cell, Vol. 9, No. 3, 2006, pp. 153-155. doi:10.1016/j.ccr.2006.02.027
- M. B. Atkins, M. S. Ernstoff, R. A. Figlin, K. T. Flaherty, D. J. George, W. G. Kaelin Jr., E. D. Kwon, T. A. Libermann, W. M. Linehan, D. F. McDermott, A. C. Ochoa, A. J. Pantuck, B. I. Rini, M. A. Rosen, J. A. Sosman, V. P. Sukhatme, J. W. Vieweg, C. G. Wood and L. King, “Innovations and Challenges in Renal Cell Carcinoma: Summary Statement from the Second Cambridge Conference,” Clinical Cancer Research, Vol. 13, No. 2, 2007, pp. 667s-670s.
- J. Brugarolas, “Molecular Pathways and Targeted Therapies for Renal Cell Carcinom,” ASCO 2009, Educational Book 2009, pp. 710-715.
- GenomeNet, “Kyoto Encyclopedia of Genes and Genomes (KEGG),” http://www.genome.jp/kegg/kegg1.html
- R. T. Kurmasheva, S. Huang and P. J. Houghton, “Predicted mechanisms of resistance to mTOR inhibitors,” British Journal of Cancer, Vol. 95, No. 8, 2006, pp. 955-960. doi:10.1038/sj.bjc.6603353
- K. E. O’Reilly, F. Rojo, Q. B. She, D. Solit, G. B. Mills, D. Smith, H. Lane, F. Hofmann, D. J. Hicklin, D. L. Ludwig, J. Baselga and N. Rosen, “mTOR Inhibition Induces Upstream Receptor Tyrosine Kinase Signaling and Activates Akt,” Cancer Research, Vol. 66, No. 3, 2006, pp. 1500-1508. doi:10.1158/0008-5472.CAN-05-2925
- M. A. Arap, A. G. Kann, G. S. Fernandes, A. C. Buzaid, S. Arap and F. C. Maluf, “New Directions in the Management of Renal Cell Carcinoma,” Cancer Therapy, Vol. 6, 2008, pp. 11-24.
- E. J. Abel and C. G. Wood, “Cytoreductive Nephrectomy for Metastatic RCC in the Era of Targeted Therapy,” Nature Reviews Urology, Vol. 6, No. 7, 2009, pp. 375-383. doi:10.1038/nrurol.2009.102
- R. A. Figlin, “Sequencing of Drugs in the Treatment of Kidney Cancer,” 2009. http://cme.medscape.com/viewarticle/702103_print
- B. I. Rini, “ASCO 2009: Latest updates on renal carcinoma,” 2009. http://cme.medscape.com/viewarticle/706713_print
- B. I. Rini, G. Wilding, G. Hudes, W. M. Stadler, S. Kim, J. Tarazi, B. Rosbrook, P. C. Trask, L. Wood and J. P. Dutcher, “Phase II Study of Axitinib in SorafenibRefractory Metastatic Renal Cell Carcinoma,” Journal of Clinical Oncology, Vol. 27, No. 27, 2009, pp. 4462-4468. doi:10.1200/JCO.2008.21.7034
- W. M. Stadler, “Effective Therapy for Metastatic Renal Cancer, Whither to Now,” Journal of Clinical Oncology, Vol. 27, No. 22, 2009, pp. 3573-3574. doi:10.1200/JCO.2009.22.5250
- Y. Je, F. A. Schutz and T. K. Choueiri, “Risk of Bleeding with Vascular Endothelial Growth Factor Receptor Tyrosine-Kinase Inhibitors Sunitinib and Sorafenib: A Systematic Review and Meta-Analysis of Clinical Trials,” The Lancet Oncology, Vol. 10, No. 10, 2009, pp. 967-974. doi:10.1016/S1470-2045(09)70222-0
- S. R. Nalluri, D. Chu, R. Keresztes, X. Zhu and S. Wu, “Risk of Venous Thromboembolism with the Angiogenesis Inhibitor Bevacizumab in Cancer Patients: A Meta-Analysis,” The Journal of the American Medical Association, Vol. 300, No. 19, 2008, pp. 2277-2285. doi:10.1001/jama.2008.656