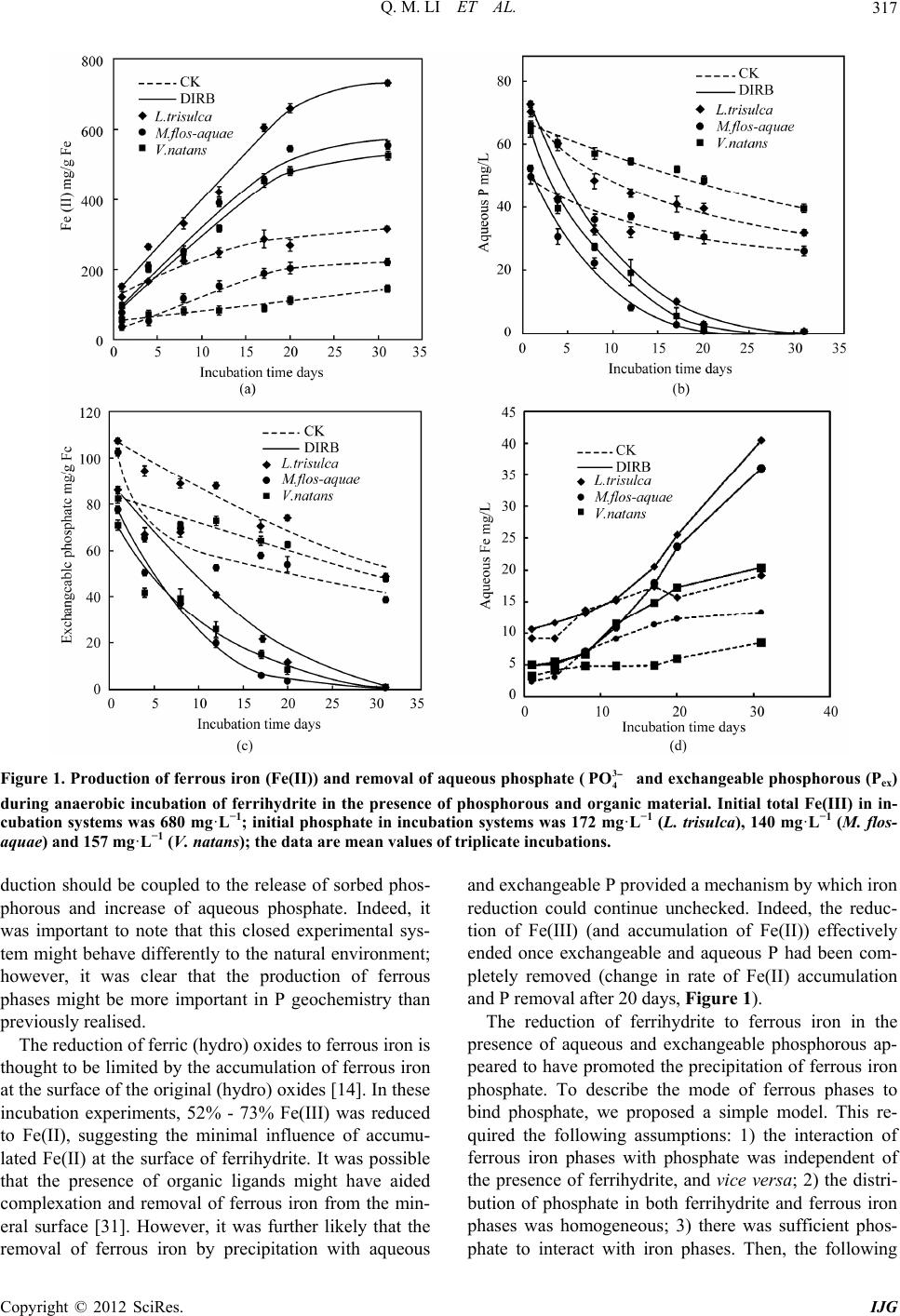
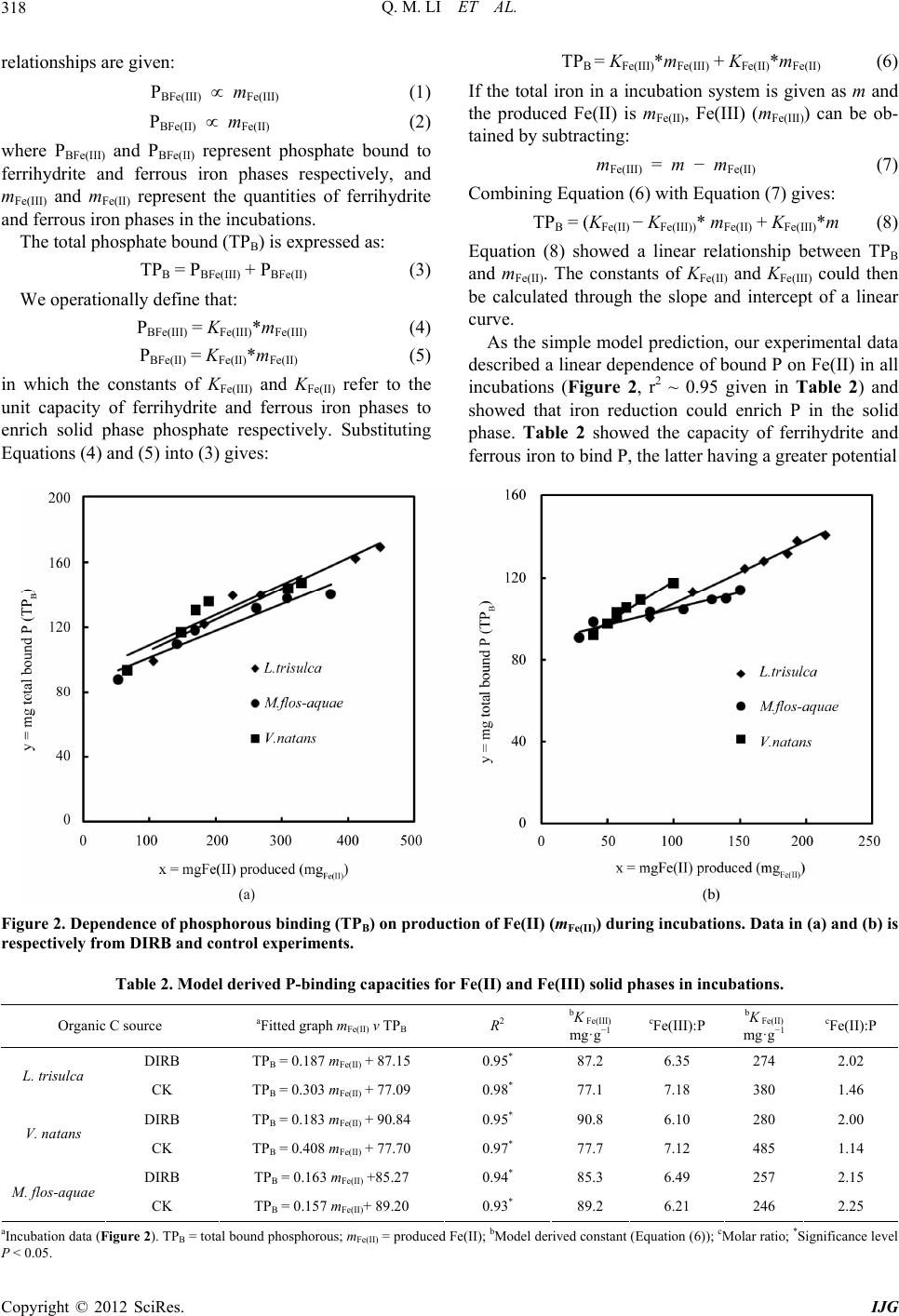
Q. M. LI ET AL.
320
doi:10.1007/s10533-005-2343-3
[8] R. A. Royer, W. D. Burgos, A. S. Fisher, B. H. Jeon, R. F.
Unz and B. A. Dempsey, “Enhancement of Hematite Bio-
reduction by Natural Organic Matter,” Environmental
Science & Technology, Vol. 36, No. 13, 2002, pp. 2897-
2904. doi:10.1021/es015735y
[9] T. S. Peretyazhko, J. M. Zachara, D. W. Kennedy, J. K.
Fredrickson, B. W. Arey, J. P. McKinley, C. M. Wang, A.
C. Dohnalkova and Y. Xia, “Ferrous Phosphate Surface
Precipitates Resulting from the Reduction of Intragrain
6-Line Ferrihydrite by Shewanella Oneidensis MR-1,”
Geochimica et Cosmochimica Acta, Vol. 74, No. 13, 2010,
pp. 3751-3767. doi:10.1016/j.gca.2010.04.008
[10] D. R. Lovley, E. J. P. Phillips and D. J. Lonergan, “En-
zymatic versus Nonenzymatic Mechanisms for Fe(III)
Reduction in Aquatic Sediments,” Environmental Science
& Technology, Vol. 25, No. 6, 1991, pp. 1062-1067.
doi:10.1021/es00018a007
[11] R. A. Royer, W. D. Burgos, A. S. Fisher, R. F. Unz and B.
A. Dempsey, “Enhancement of Biological Reduction of
Hematite by Electron Shuttling and Fe(II) Complexa-
tion,” Environmental Science & Technology, Vol. 36, No.
9, 2002, pp. 1939-1946. doi:10.1021/es011139s
[12] R. A. Doong and B. Schink, “Cysteine-Mediated Reduc-
tive Dissolution of Poorly Crystalline Iron(III) Oxides by
Geobacter Sulfurreducens,” Environmental Science &
Technology, Vol. 36, No. 13, 2002, pp. 2939-2945.
doi:10.1021/es0102235
[13] J. .K Fredrickson, S. Kota, R. K. Kukkadapu, C. Liu and J.
M. Zachara, “Influence of Electron Donor/Acceptor Con-
centrations on Hydrous Ferric Oxide (HFO) Bioreduc-
tion,” Biodegradation, Vol. 14, No. 2, 2003, pp. 91-103.
doi:10.1023/A:1024001207574
[14] R. A. Royer, B. A. Dempsey, B. H. Jeon and W. D. Bur-
gos, “Inhibition of Biological Reductive Dissolution of
Hematite by Ferrous Iron,” Environmental Science &
Technology, Vol. 38, No. 1, 2004, pp. 187-193.
doi:10.1021/es026466u
[15] E. J. O’Loughlin, “Effects of Electron Transfer Mediators
on the Bioreduction of Lepidocrocite (Gamma-FeOOH)
by Shewanella Putrefaciens CN32,” Environmental Sci-
ence & Technology, Vol. 42, No. 18, 2008, pp. 6876-
6882. doi:10.1021/es800686d
[16] S. Rakshit, M. Uchimiya and G. Sposito, “Iron(III) Bio-
reduction in Soil in the Presence of Added Humic Sub-
stances,” Soil Science Society of America Journal, Vol.
73, No. 1, 2009, pp. 65-71. doi:10.2136/sssaj2007.0418
[17] P. P. Hearn and D. L. E. C. Parkhurst, “Authigenic Vivi-
anite in Potomac River Sediments: Control by Ferric
Oxyhydroxides,” Journal of Sediment Research, Vol. 53,
No. 1, 1983, pp. 165-177.
[18] R. L. Frost, W. Martens, P. A. Williams and J. T. Klo-
progge, “Raman and Infrared Spectroscopic Study of the
Vivianite-Group Phosphates Vivianite, Baricite and Bo-
bierrite,” Mineralogical Magazine, Vol. 66, No. 6, 2002,
pp. 1063-1073. doi:10.1180/0026461026660077
[19] R. K. Kukkadapu, J. M. Zachara, J. K. Fredrickson and D.
W. Kennedy, “Biotransformation of Two-Line Silica-Fer-
rihydrite by a Dissimilatory Fe(III)-Reducing Bacterium:
Formation of Carbonate Green Rust in the Presence of
Phosphate,” Geochimica Cosmochimica Acta, Vol. 68,
No. 13, 2004, pp. 2799-2814.
doi:10.1016/j.gca.2003.12.024
[20] N. Parmar, Y. A. Gorby, T. J. Beveridge and F. G. Ferris,
“Formation of Green Rust and Immobilization of Nickel
in Response to Bacterial Reduction of Hydrous Ferric
Oxide,” Geomicrobiology of Journal, Vol. 18, No. 4,
2001, pp. 375-385. doi:10.1080/014904501753210549
[21] U. Schwertmann, D. G. Schulze and E. Murad, “Identifi-
cation of Ferrihydrite in Soils by Dissolution Kinetics,
Differential X-Ray Diffraction and Mössbauer Spectros-
copy,” Soil Science Society of America Journal, Vol. 46,
No. 4, 1982, pp. 869-875.
doi:10.2136/sssaj1982.03615995004600040040x
[22] J. .L Jambor and J. E. Dutrizac, “Occurrence and Consti-
tution of Natural and Synthetic Ferrihydrite, a Wide-
spread Iron Oxyhydroxide,” Chemical Reviews, Vol. 98,
No. 7, 1998, pp. 2549-2585. doi:10.1021/cr970105t
[23] J. C. Ryden, J. R. McLaughlin and J. K. Syers, “Mecha-
nisms of Phosphate Sorption by Soils and Hydrous Ferric
Oxide Gels,” Journal of Soil Science, Vol. 28, No. 1,
1977, pp. 72-92. doi:10.1111/j.1365-2389.1977.tb02297.x
[24] S. Kuo and E. G. Lotse, “Kinetics of Phosphate Adsorp-
tion and Desorption by Hematite and Gibbsite,” Soil Sci-
ence, Vol. 116, No. 6, 1973, pp. 400-406.
doi:10.1097/00010694-197312000-00002
[25] R. L. Parfitt and R. J. Atkinson, “Phosphate Adsorption
on Goethite (α-FeOOH),” Nature, Vol. 264, No. 5588,
1976, pp. 740-742. doi:10.1038/264740a0
[26] J. Torrent, V. Barrón and U. Schwertmann, “Phosphate
Adsorption and Desorption by Goethites Differing in
Crystal Morphology,” Soil Science Society of America
Journal, Vol. 549, No. 4, 1990, pp. 1007-1012.
doi:10.2136/sssaj1990.03615995005400040012x
[27] L. Madrid and P. D. Arambarri, “Adsorption of Phos-
phate by Two Iron Oxides in Relation to Their Porosity,”
Journal of Soil Science, Vol. 36, No. 4, 1985, pp. 523-
530. doi:10.1111/j.1365-2389.1985.tb00355.x
[28] W. R. Fischer, “Standard Potentials (E0) of Iron(III) Ox-
ides under Reducing Conditions,” Zeitschrift fuer Pflan-
zenernaehrung und Bodenkunde, Vol. 150, No. 5, 2007,
pp. 286-289. doi:10.1002/jpln.19871500504
[29] R. J. Atkinson, A. M. Posner and J. P. Quirk, “Crystal
Nucleation in Fe(III) Solutions and Hydroxide Gels,”
Journal of Inorganic and Nuclear Chemistry, Vol. 30, No.
9, 1968, pp. 2371-238.
doi:10.1016/0022-1902(68)80247-7
[30] D. R. Lovley and E. J. P. Phillips, “Novel Mode of Mi-
crobial Energy Metabolism: Organic Carbon Oxidation
Coupled to Dissimilatory Reduction of Iron or Manga-
nese,” Applied and Environment Microbiology, Vol. 54,
No. 6, 1988, pp. 1472-1480.
[31] W. Zhang, Q. M. Li, X. X. Wang, Y. Ding and J. Sun,
“Reducing Organic Substances from Anaerobic Decom-
position of Hydrophytes,” Biogeochemistry, Vol. 94, No.
1, 2009, pp. 1-11. doi:10.1007/s10533-009-9295-y
Copyright © 2012 SciRes. IJG