Evaluation Procedure for Quality Consistency of Generic Nifedipine Extended-Release Tablets Based on the Impurity Profile ()
1. Introduction
Generic drug is a kind of imitation that owns the same dosage, safety, effectiveness, quality and indication with the original drug. However, the generic products are commonly inconsistent with the original product, especially in quality. For example, some new impurities or higher content of impurities can be found in generic drugs compared with the original product [1] [2] . The reason is that generic drugs can be listed as long as they accord with the relevant quality standards, even though their quality falls short of the original drug. But these quality standards may only limit the total impurities, not the number of impurities. The presence of certain impurities can lead to a decrease or alteration of biological activity, or even impact the safety of the drug. For instance, the alteration of the production process of tryptophan from chemical synthesis to fermentation led to the formation of new impurities, which was the reason of a severe side effect, eosinophila-myalgia syndrome, causing the death of 27 patients [3] . In 2000, it was reported that 66 gentamicin patients died, because of different impurities in bulk drugs from various manufacturers [4] [5] .
The definition of the impurity profile was given in the guidelines of ICH in 2002, which was stated as: a description of the identified and unidentified impurities, present in a new drug substance [6] [7] . The focus not only limits one or some impurities, but also traces the formation and variation of the whole impurities. It can help the researchers to control the drug quality in synthesis, process, storage, circulation and so on. High quality generic drugs should be equivalent to the original drug in biology, clinic, and safety. If generic drugs can be equal to (if not better than) the original product in the number and the amount of impurities, they should be consistent in safety with the original product to a certain extent, which also provides the basis for biological and clinical equivalence. At present, the quality consistency of generic drugs in some countries was evaluated by the drug release rate and clinical effects, while impurity profile could be a new perspective for evaluation.
A few investigations based on impurity profile combined with chemometrics have been reported in the literature. M. Dumarey et al. [8] performed PCA, projection pursuit (PP), HCA and auto-associative multivariate regression trees (AAMRT) to recognize paracetamol via impurities profile. The methods used PCA, Fisher-ratio, degree-of-class separation (DCS), KNN (K-nearest neighbors), and also combined with impurities profile of eight KCN (potassium cyanide) stocks. These methods were confirmed as useful to prevent crimes and terrorist attacks [9] - [11] . There are some other cases, for instance, the HCA is used to recognize the same batch via impurities profiles of methamphetamine, so that the source of drugs can be traced [12] . However, there is still no report which uses impurity profile combined with chemometrics to evaluate the quality consistency of generic drugs.
In this study, 6 copies produced in China, containing nifedipine are compared to the original drug (Adalat® retard) for the uniformity of impurity profile. Then, a strategy is outlined for evaluating the similarity between generic drugs and the original drug. This is accomplished with the aid of the chemometrics methods which are used for calculating the similarity of chromatographic fingerprint for herbal medicine [13] . Knowledge gained from this investigation provides the basis and a new idea for evaluating the quality consistency of generic drugs.
2. Theory
The following sections describe the algorithms utilized in this paper.
2.1. Cosine and Correlation Coefficient
Cosine and correlation coefficient were two classical methods for evaluating the similarity between two chromatograms. Chromatogram can be treated as vector of hyperspace, and the similarity between them can be counted according to angle cosine formula and correlation coefficient formula. The more near to 1.0 the value of cosθ or R is, the more similar are the two vectors. Similarities of two chromatograms were expressed as cosine formula (cosθ).
 (1)
(1)
And correlation coefficient formula (R)
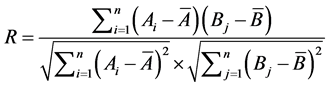 (2)
(2)
In this paper, Ai, is the ith variate of the chromatogram of original drug, and Bj is the ith variate of chromatogram of general drugs, and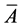 ,
,  are the mean values of them, respectively. n is the number of variates [14] .
are the mean values of them, respectively. n is the number of variates [14] .
2.2. Adaptive Iteratively Reweighted Penalized Least Squares (AirPLS)
The airPLS algorithm is a simple but flexible, valid and fast algorithm for estimating baselines in analytical chemistry. There is one crucial but intuitional parameter λ to control the smoothness of the fitted baseline. It gives extremely fast and accurate baseline corrected signals for both simulated and real signals. The approach has been applied to chromatograms, Raman spectra and NMR signals [15] .
2.3. Correlation Optimized Warping (COW)
The approach for aligning signals, termed correlation optimized warping (COW), was proposed in 1998 as a means to correct chromatograms for shifts in the time axis prior to multivariate modeling. It is a piecewise or segmented data preprocessing method (operating on one sample record at a time) aimed at aligning a sample data vector towards a reference vector by allowing limited changes in segments lengths on the sample vector [16] [17] .
2.4. Principal Component Analysis (PCA) and Hierarchical Cluster Analysis (HCA)
HCA and PCA are two unsupervised pattern recognition techniques widely used in chemometrics [18] . PCA is a variable reduction method, which projects multivariate data into a lower-dimensional space. This is achieved by determining latent variables, principal components (PC’s), which maximize description of the data variation. Depend on them, visualization of the data is achieved and possible clustering tendencies can be observed by plotting the scores of one PC versus the scores of another selected PC [19] . In HCA, the most similar objects are sequentially merged in clusters until one big cluster is obtained. The higher two objects are connected in the dendrogram, the more dissimilar they are [20] [21] .
2.5. Weighted Correlation Coefficient
Weighted correlation coefficient was used for further evaluation of the generic quality. Similarities of two chromatograms were expressed as weighted correlation coefficient formula (R')
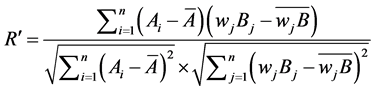 (3)
(3)
 is the weight of ith peak area in chromatograms of the generic drugs.
is the weight of ith peak area in chromatograms of the generic drugs.
3. Experimental
3.1. Reagents and Samples
Nifedipine, nifedipine impurity I and nifedipine impurity II which were used as reference substances were obtained from the National Institute for Food and Drug Central (Beijing, China). The generic drugs (20 mg) from 6 factories with various batches were named as B1, B2, B3, C1, D1, E1, E2, E3, F1, F2, F3, G1, G2, G3, and the negative control drug (Amikacin sulfate infection) was named as N1, which were purchased from some local pharmacies in China. The original products of nifedipine extended-release tablets, Adalat® retard, which were named as A1 and A2, were purchased from Bayer Health Care AG (Leverkusen, German). All adjuvants include: lactose, hypromellose K4, povidone K30, magnesium stearate were obtained from the department of pharmaceutical sciences in second military medicine university. The HPLC grade acetonitrile, methanol and chloroform were purchased from Merck (Darmstadt, Germany).
3.2. Forced Degradation Studies
The samples (bulk drugs, excipients and preparations) were subjected to accelerated degradation conditions including acid, alkaline, oxidative, photolytic, and thermal stress, to observe the changes of degradation products. Samples prepared in 1 mol/L hydrochloric acid were used for the acidic hydrolysis, and samples in 1 mol/L sodium hydroxide for the basic hydrolysis evaluation. Both samples were kept at ambient temperature for 22 hours before neutralizing with base or acid. The oxidative degradation was induced by storing the samples in 15% hydrogen peroxide, at ambient temperature for 22.5 hours in dark place. Photodegradation was induced by exposing the samples to 4500 lx near ultraviolet light for 1 hour. Thermal degradation was induced by heating the samples at 105˚C for 4 hours. Multiple stressed samples were chromatographed along with a non-stressed sample.
3.3. Chromatographic Measurement
15 batches of samples were analyzed according to the IMPURITIES in USP-34 [22] . The chromatographic separation was performed on Agilent 1200 HPLC system (Agilent Technologies, CA, USA) with a variable wavelength detector. The mobile phase consisted of mixture of acetonitrile, methanol and water (25:25:50, v/v/v). The column is a phenomenex Luna 5u C18(2) 100 A 250 × 460 mm, 5micron. The flow rate was 1 ml/min. A 25 μL volume of each sample was injected and the chromatogram was monitored at a wavelength of 265 nm.
Three individual preparations were made for all batches, which were used for evaluating the consistency of generic drugs. Six individual preparations were made for batch: A1, B1, C1, D1, E1, F1, and G1, which were also used for evaluation of intra-batch similarity. The measurements were completed in a continuous time and the peak areas were recorded by automatic integration.
3.4. Data Processing
Correlation coefficient and weighted correlation coefficient were calculated with programs written in Matlab R
2008a
(the Mathworks, USA). Cosine and HCA were performed in PASW 18.0 (IBM, USA). PCA was performed in SIMCA 11.5 (Umetrics, Sweden).
4. Results and Discussion
4.1. Choice of Chromatographic Methods
The method for collecting the data of impurity profile is the basis for the quality consistency evaluation of generic drugs. It can be chosen from the pharmacopoeias or quality standards of drugs in every country or organization, or even a new but validated method. The parameters like columns and reagents from different factories also should be investigated in the same standard. The criterion is the method has the ability to detect more impurities peaks which meet the requirements of quantitative analysis. In this study, two standards were collected. One is USP-34 [22] ; the other is National Drug Standard [23] issued by China’s State Food and Drug Administration. Finally, the method of USP-34 was chosen to collect the date, which was based on the consideration that: 1) the number, separation and plate number of impurities which can be detected met the requirement of the analysis; 2) the method was more convenient and safer; 3) the excipients and reagents have no interference to the detection of impurities.
4.2. Data Collection and Conformity Test
Before further calculation, there is a need to test whether the content and impurities of each batch conform to the quality standard (conformity test). The results showed that the content of specified impurities (impurity I and impurity II) and total impurities in all samples were qualified. However, as can be seen from the chromatograms (Figure 1), the number and/or peak areas of unspecified impurities in the generic products were higher than those of the original product. Therefore, it cannot completely evaluate the generic level of generic drugs by only limiting the content of one or some impurities.
The chromatographic data of conformity test were also used for the following evaluation.
![]()
Figure 1. The chromatogram of each factory.
4.3. Evaluation of Intra-Batch Similarity of Drugs
The qualified samples can be subsequently evaluated for their intra-batch similarities. It was expected that the impurity profile generated from the same batch will be similar under the same analytical condition. So, when the similarity of intra-batch samples was higher, the process would be more stable and the following comparison with the original drug would be more significant. In this study, the intra-batch differences turned into the comparison of similarity coefficient by using correlation coefficient and cosine. Referring to two kinds of vectors for calculating the similarity in chromatographic fingerprint for herbal medicine, the results of two kinds of vectors were also compared: one is data point of the whole chromatogram (point-by-point) and the other is peak area (peak-by-peak).
4.3.1. Data Point (Point-by-Point)
In chromatogram of chemical drugs, the peak of main compound was thousand times more intense than those of the impurities. The information of these impurities might be leveled by the main compound when evaluating the similarity of intra-batch samples, so there is a need to remove 200 data points of main compound from the initial data. In order to simplify the calculation, the data from 0 - 10 min, 32 - 60 min, which belonged to excipients peaks and baseline, were also removed.
Because fluctuation of baseline was apparent when data was collected, Adaptive Iteratively Reweighted Penalized Least Squares (airPLS) [15] [24] and correlation optimized warping (COW) [17] were used to correct the baseline and peak shift which was caused by instrumental variability. The results of two methods showed that there was a big difference in intra-batch samples when using initial data. For G1, the range of correlation coefficient and cosine before correction were 0.1230 - 0.9578 and 0.0077 - 0.9212, respectively. The corresponding values after correcting were 0.7954 - 0.9741 and 0.6966 - 0.9528, respectively.
Although the instrument has been balanced for more than 2 hours, baseline drift was still presented. This may be closely related to the column and mobile phase. The intra-batch similarity was significantly increased after correcting, because it can partly eliminate the influence of baseline and peak drift caused by environmental disturbance. However, the result was still not satisfactory.
4.3.2. Peak Area (Peak-by-Peak)
The peak areas were recorded by automatic integration. Both the correlation coefficient and cosine obtained the uniform results, showing that the samples were similar in the same batch. The similarity results of G1 were 0.971 - 0.999 and 0.964 - 1, respectively.
When peak areas were used to calculate the similarity of intra-batch samples, a reasonable result (mean similarity of all factories > 0.9) was obtained. It revealed that the process of each manufactory was stable. Because there is no need to export, cut or correct data, it was more convenient to use the peak areas as vector if the peaks were separated well. The following evaluations were all based on calculation of the peak areas, instead of the data point. All the qualified generic products were then sent to the following evaluation step.
4.4. Evaluation of Generic Drugs with the Original Drug
To meet the requirement of following calculation, when a special impurity at certain retention time emerged in one batch which was absent in the other batches, the absent peak area was recorded as 0.00001.
4.4.1. PCA
The PCA calculation was based on a singular value decomposition of the data array of the impurity profile. The first two scores of PCA results (Figure 2) were used to make projection plot that provide a visual determination of the similarity among the samples. It was considered that the more similar with the original drug in impurities, the closer the generic drug was to A
1 in
Figure 2.
The resulting PC1-PC2 score plot made a clear distinction between the N1 and the test samples. The results showed the differences among the batches and factories. The Figure 2 presented that samples from D, F and B2 were far from A1, A2, which meant there were some differences among them. The whole batches from C, E, G clustered to A1, A2, which indicated they were more similar to the original drug. When applying PCA on this dataset, the amounts of variance explained by the first five PC’s are 48.36, 22.13, 12.60, 8.36, and 5.94, respectively. However, our goal, which was to pick out the best generic factory, was not clearly achieved.
![]()
Figure 2. PCA scores of the seven manufactories.
4.4.2. HCA
The peak areas data were subjected to the Ward hierarchical clustering method using the Euclidean distance as a similarity measure, resulting in the dendrogram presented in Figure 3. The faster connected with A1, A
2 in
the dendrogram, the more similar the generic drugs are to the original drug.
![]()
Figure 3. HCA dendrogram of the seven manufactories.
All samples in the dendrogram, except for the E3-3, formed a subgroup with samples from the same batch. The N samples did not cluster themselves with the other Nifedipine samples. All samples from the G and A (original samples) formed a subgroup and then linked to the other samples. However, as the same with PCA, the samples from the same factory were not clearly separated from the other samples.
For their graphic form of presentation of the result, PCA and HCA can intuitively provide the differences among inter-batches and generic level of the generic products. They can help a pre-judgment for appraiser and generic factories to evaluate and improve the quality of generic drugs. F was considered as an obvious outlier and in need of improving the manufacturing process. Whether the other generic drugs are similar to the original drug is still ambiguous based only on PCA and HCA results, so they were sent to further investigation except F.
4.5. Weighted Similarity Evaluation of Generic Drugs
Generally speaking, the specified impurities were paid more attentions in quality control for their higher content. But for generic drugs, the unspecified impurities especially the special impurities or process impurities were commonly the presentation of different manufacturing process. However, their information was easily concealed by the impurities which have higher concentration (usually specified impurities) in similarity calculation. To evaluate the contribution of these unspecified impurities, the preparations, APIs, and excipients of E2 were subjected to forced degradation test to observe the change of each impurity. The detections were not interfered by excipients and the impurities were well-separated after the forced degradation test. Impurity I and II had undergone the most obvious changes in the test, which were the specified impurities in quality standard of nifedipine extended-release tablets in USP-34. The last 4 impurities (Figure 1) were confirmed to be the process impurities because they had no obvious change in the test. Among the other samples, the areas of these process impurities were much smaller than the specified impurities.
In order to improve the contribution of “the unspecified impurities” in similarity calculation, the “weight” was introduced based on the perspective of impurities profile. However, to avoid excessive weighting, the upper limit (S) of weight was defined as: S = Cmin/Pmax, where Cmin corresponds to the mean peak areas of the smallest specified impurity in the original drug, Pmax corresponds to the mean peak areas of the biggest unspecified impurity in the original drug. S equals to 2.8, a moderate value, in this case of nifedipine extended-release tablets.
Figure 4 presented the weighted correlation coefficient results. Before weighting, the similarity of D and G were close. After weighting, the similarity of the generics all decreased. The decline of G was the smallest, from 0.84 to 0.83, while the biggest drop of the mean similarity was D, from 0.85 to 0.66. The reasons can be derived from the chromatograms (Figure 1). Special impurity appeared in B, C, and D factories leading to their plummeted similarities, so they should be excluded from the evaluation procedure. The concentrations of unspecified impurities in E, although whose impurity I and impurity II were well controlled, were higher than those of A, so process of E also need further improvement. The differences between the four generic drugs and the original drug led to different degrees of similarity decrease. Via adding weight to the unspecified impurities, the generic level could be further evaluated.
Although the impurities peak areas of G were larger than the original drug, it was the smallest of all the generic drugs in this paper. Hence, G was considered to have the (comparatively) best generic quality for its intra-batch high similarity and the highest weighted similarity to the original drug.
A process chart (see Figure 5) details the evaluation procedure for quality consistency of impurity profile in generic drugs, where a step-by-step guide facilitates the estimation of consistency between the generic drugs and the original drugs.
5. Conclusion
The purpose of this study is to present an evaluation process and recommendations for estimating the quality consistency of generic drugs, taking 6 copies of nifedipine extended-release tablets as examples. The similarity analysis methods in chromatographic fingerprint for herbal medicine and impurity profiles were combined to accomplish the evaluation procedure. Sequentially, correlation coefficient, cosine can quantify the similarity of intra-batch drugs. The differences of inter-batch products and generic level are visualized by PCA and HCA. The generic level can be further evaluated by weighting the unspecified impurities. While future efforts are still needed to explore the structure and the effect of unidentified impurities, this study successfully establishes an evaluation process and can expect a widespread application in the all-around evaluation of generic drugs.
![]()
Figure 4. The change of similarity coefficient before and after weighting in A, B, C, D, E, G factories.
![]()
Figure 5. Evaluation flowchart of quality consistency based on impurity profile of the generic drugs.
NOTES
*These authors contributed equally to this work.
#Corresponding author.