1. Introduction
People living with diabetes would reach 366 Vs 171 million by 2030, out of which 90 to 95% are type 2 diabetic patients. Apart from chronic diabetic related problems such as nephropathy, neuropathy and accelerated atherosclerosis, the following groups of people with diabetics and HIV can be identified: 1) patients with preexisting diabetics contact HIV; 2) patients who are diagnosed to have diabetics at the onset of HIV; 3) HIV patients diagnosed with gestational diabetics; 4) patients who develop diabetics after HIV therapy [1] .
Patients with HIV and diabetics would be on polytherapy and these subjects would be more prone to DDIs. HIV drugs notably PIs are potent inhibitors of drug metabolizing enzymes and drug transporters. Majority of PIs showed irreversible inhibition of CYP3A4 which is major metabolizing enzyme in human contributing to metabolism of about 50% of marked drugs [2] . In a previous study in our lab we had seen that except indinavir all tested PIs demonstrated irreversible inhibition of CYP3A4 (Ritonavir, Indinavir, Nelfinavir, Saquinavir and Amprenavir) and these results were in agreement with published results [3] . Many of the anti-HIV drugs like RTV are also OATP inhibitors [4] .
Pharmacokinetic drug interactions were observed in repaglinide with drugs which are inhibitors of CYP3A4, CYP2C8 and transporter OATP1B1. The DDIs were severe in case of CYP2C8 and OATP1B1. However the inhibitions were severe in case of CYP inhibitions than that in transporters inhibitions. More than double repaglinide AUC (144%) was noticed when coadministered with cyclosporine, a potent OATP1B1 inhibitor. Severe DDIs were observed when gemfibrozil was coadministered with repaglinide; an eight fold increase in repaglinide AUC was seen. This drug interaction was further enhanced with introduction of itraconazole. OATP inhibition seems to be involved only at higher doses. In a separate study subtherapeutic doses (<300 mg) of gemfibrozil in healthy volunteers caused mechanism based inhibition by formation of metabolite (gemfibrozil 1-O-β-glucuro- nide) with higher levels of repaglinide [5] .
Furthermore, AUC of repaglinide was higher with subjects with higher activity of CYP2C8*1/*3 genotype than with lower activity of CYP2C8*1/*1 genotype. According to EMEA’s (committee for Proprietary Medicinal Products) using this combination was declared as contraindication [5] . On the other hand selective inhibitor of CYP2C8 by trimethoprim increases the AUC of repaglinide by 61% in healthy subjects [5] .
Rifampicin a antitubercular drugs which is prototype inducer of CYP3A4 and OATP inhibitor, interfered the pharmacokinetics of repaglinide. The exposure of repaglinide is influenced by rifampicin treatment for example repaglinide AUC was decreased by 31% when repaglinide is treated one hour after the last dose of rifampicin. In a separate study, AUC of repaglinide was decreased to 57% when ingested 12.5 hours after last dose rifampicin [6] . Yet in another study AUC of repaglinide is affected by 50% and 80% when repaglinide administrated concomitantly and 24 hours after the last treatment of rifampicin respectively [6] . From this study we can hypothesize that ripaglinide disposition would be effected by OATP transporter, which makes repaglinide available for metabolism and CYP3A4 which plays a role in metabolism. Influencing CYP3A4 and OATP would alter the PK of the repaglinide. HIV drugs which are known CYP3A4 inhibitors and OATP inhibitors would alter pharmacokinetics of repaglinide. http://www.hiv-druginteractions.org published by liver pool university indicated some possible DDIs of anti-HIV drugs when repaglinide was coadministrated. They indicated that close monitoring or dose adjustment was required when repaglinide dosed with atazanavir, darunavir, fosamprenavir, indinavir, lopinavir, nelfinavir, ritonavir, saquinavir and tipranavir.
In the current study we evaluated the effect of repaglinide metabolism in HLM and MLM, furthermore tested the mechanism based inhibition in HLM and MLM. RTV and KTZ were also tested for DDIs in mouse model. KTZ was used as positive control for CYP3A4 inhibition in in vitro and in vivo.
2. Materials and Methods
Drugs and Chemicals. Midazolam, ketoconazole, ritonavir, amprenavir, indinavir, nefinavir, Maraviroc, Delavirdine and Nevirapine purchased from Kemprotec (United Kingdom). MLM XenoTech, LLC (Kansas City). Verapamil, repaglinide, polyethylene glycol 400, NADPH from Sigma-Aldrich (St Lous, MO, USA) nateglinide, efavirenz gift from Dr. Reddy’s and Hetero Drugs (Hyderabad, India) respectively. All the chemicals used were HPLC grade.
Animals. In vivo mouse studies were performed according to the Guidelines for the Care and Use of Laboratory Animals that was approved by the Committee of Ethics of Animal Experimentation of J.S.S college of Pharmacy, Ooty. 8 to 12 weeks old Female Balb/c mice used in the same facility. Animals were housed in a temperature- and humidity-controlled room with a 12-h light/dark cycle. Animals were fed with standard animal diet (Harlan mice diet); food and water was provided ad libitum. Mice were fasted overnight before oral dose and fed after 2 hours of dosing, for multiple dosing mice were fasted on the last dose.
Enzyme Kinetics (HLM & MLM). To determine the Km and Vmax values (the apparent enzyme kinetic constants), repaglinide was incubated in duplicates with an incubation mixture containing HLM or MLM with 0.2 mg/ml protein (repaglinide concentration 0.334, 1, 3, 9, 27, 81, 243 µM) and MgCl2 (3.3 mM) in 100 mM potassium phosphate buffer pH 7.4, after 5 mins equilibration in water bath at 37˚C the reaction was started with NADPH (NADPH (1.3 mM), 0.2 ml total volume). The reaction was terminated with acetonitrile containing internal standard (nateglinide 500 ng/ml). The samples were stored at −80˚C and analyzed later. All the incubations were accepted only if midazolam metabolism was >90%.
IC50 & Time Dependent IC50. Incubation conditions were similar to the enzyme kinetic experiment, unless otherwise specified. Repaglinide concentration was tested at the Km value for both HLM and MLM, inhibitor concentrations for APV were (50, 16.665, 5.555, 1.851, 0.615, 0.205 µM), RTV and KTZ (10, 3.334, 1.111, 0.370, 0.123, 0.041, 0.013, 0.004 µM). The reaction was terminated after 30 minutes of incubation and incubation accepted as in the Km experiment non dilution method was used for time dependent IC50 (repaglinide at Km concentration), inactivators were incubated along with NADPH for 30 mins, after which substrate was added and incubated for 5 mins [7] [8] . This was answered in enzyme kinetics. After the incubation samples were stored at −80˚C and analyzed later, the incubation pass criteria was as specified earlier.
In vivo studies. Oral dosing formulation for repaglinide and inhibitors were formulated in 10% Ethanol: 60% PEG400: 30%: 5% dextrose in water (D5W). With these cocktail vehicles all the drugs were in solution state. The weighed drugs were transferred to mortar and pestle, vehicles were added in following order, ethanol, PEG 400 and D5W. After adding ethanol compound was triturated followed by PEG 400 and D5W. All the formulations were made fresh before dosing. Repaglinide was dosed 1 hour after inhibitor dosing, sampling time was started after repaglinide dosing. Blood samples were centrifuged to separate plasma and analyzed using HPLC- mass spectrometry. Composite sampling is taken from retro orbital. After dosing of repaglinide the following time points were taken 0, 0.5, 1, 2, 4, 8 and 24 hours. The collected samples were stored at −80˚C until analysis.
Analytical Methods. All sample analysis was carried out using high performance liquid chromatography (HPLC)―tandem mass spectrometry methods. The HPLC elute was introduced via electrospray ionization. Mass spectral analysis was performed using multiple reaction monitoring with the following transitions (positive ionization mode) m/z amprenavir 506.4 → 155.7, indianvir 614.5 → 421.3, nelfinavir 568.1 → 330.1, saquinavir 671.9 → 570.5, ritonavir 721.9 → 296.3, ketoconazole 531.1 → 82, efavirenz 316.2 → 244.2, repaglinide 453 → 230.4 and nateglinide 318.3 → 125.
The mass spectrometry 3200 QTRAP (ABI Sciex, Applied Biosystems) connected with HPLC Agilent 1100 MSD system (Agilent Technologies, Palo Alto, CA) and auto sampler CTC PAL (Leap Technologies, Carrboro, NC). Chromatography was performed by using Phenomenex Luna C-18 column (particle size 3 µm, 50 × 2 mm) preceded by Phenomenex C-18 guard column (Phenomenex, Torrance, CA), column temperature was set to 40˚C, injection volume 10 µL. Mobile phase consisting of 10mM ammonium formate in 0.1% formic acid with acetonitrile with a flow rate 500 µL for in vitro and in vivo samples. The run time and gradient (ACETONI- TRILE %) as follows 0 min to 0.5 min 70%, 0.5 min to 2.5 min 30%, 2.5 min to 2.6 min 2%, 2.6 min to 3.2 min 100% and 3.2 min to 4 min 100%.
Sample Preparation. In vitro samples were precipitated with acetonitrile containing IS (nateglinide) at 500 ng/ml, centrifuged and injected into the LC-MS/MS. The plasma samples were extracted using liquid-liquid extraction method. In brief to 20 µl plasma sample 1 ml of methyl tertiary butyl ether is added and shaken for 10 min, freezed and supernatant is dried and reconstituted with 200 µl water methanol 1:1. 10 µl of sample is injected into LC-MS/MS.
3. Data Analysis
In vitro Data Analysis
Enzyme kinetics for repaglinide and midazolam were estimated by standard equation using GraphPad Prism software equation:
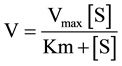
IC50 and MBI IC50 were determined by nonlinear regression analysis with GraphPad Prism software (version 5, GraphPad Software Inc., SanDiego).
To determine the enzyme inactivation kinetics constant for Human and mouse liver microsomes, the natural logarithm of remaining OH-midazolam and OH-repaglinide is plotted against the pre-incubation time incubated with inhibitors.
Pharmacokinetic Analysis. In vivo data analysis was carried out with noncompartmental analysis by using WinNonlin Professional (version 4.0.1; Pharsight, Mountain View, CA). In brief, the Cmax and Tmax were recorded by visual observation of the data. The area under the plasma concentration-time curve (AUC tot) was calculated using linear and log trapezoidal summations.
Statistical Analysis. The statistical significance (p < 0.05) between treated (repaglinide dosed with perpetrators) and control (repaglinide alone) groups were established by Dunnett’s multiple comparison test.
4. Results and Discussions
The tested HIV drugs are well known CYP3A4 inhibitors and most of them are irreversible inhibitors, notably the PIs for example, AMP, Nelfinavir, RTV, Saquinavir and Delavirdine [3] . To test possible hits we screened inhibitors at 50 µM and incubated with one µM of repaglinide, incubation conditions were described as in method described in Km and Vmax section. Out of in vitro screens in HLM and MLM we identified AMP, KTZ, Efavirenz and RTV demonstrated inhibition of metabolism of repaglinide. To further test these hits Km and Vmax values were generated in HLM and MLM (Figure 1(a) & Figure 1(b)). Furthermore the repaglinide is incubated at Km with different concentration of inhibitors, we have observed inhibitions at top 3 concentrations of inhibitors for KTZ, AMP and RTV because of which we could not generate exact IC-50 values. RTV and AMP are known mechanism based inhibitors (MBI); these two compounds were further evaluated for MBI’s along with KTZ which is included as negative control. In MBI (time dependent inhibitions) we have not noticed differences in inhibitions when compared with reversible inhibitions.
The in vitro hits tested in in vivo using mouse as an animal model. The DDIs at 0.1 mg/kg of repaglinide and RTV and KTZ (doses 45 and 40 mg/kg respectively, dose was normalized to human plasma concentrations) (Figure 2(a) & Figure 2(b)) were so strong that mice were either moribund or dead. The dose of repaglinide was reduced to 5 fold (0.02 mg/kg) and dosed, same kind of toxicity is observed. Noticing strong DDIs the dose of repaglinide is reduced to 20 fold (0.005 mg/kg), where we did not observe any adverse effects (the cage behavior was normal). This dose was used to for repaglinide to evaluated pharmacokinetics DDIs with KTZ and RTV. Repaglinide dosed with KTZ in mouse, the Cmax of repaglinide increased from undetectable levels (less than 2 ng/ml) to 11.63 ng/ml, which could be due to CYP3A4 inhibition. Similarly in the clinic KTZ and clarithromycin which are strong CYP3A4 inhibitors increased the Cmax of repaglinide significantly with less effect on AUC, indicating that CYP2C8 would take over the metabolism when CYP3A4 is inhibited [5] . In a separate study when CYP3A4 induced and OATP inhibited by rifampicin the DDI were stronger than inhibition CYP3A4 alone. This is further strengthened by potent inhibition of gemfibrozil which is CYP2C8 irreversible inhibitor and also inhibitor of OATP transporter. Gemfibrozil DDI indicates that the repaglinide Pharmacokinetics will be altered greatly with interfering with CYP2C8 and influx transporter OATP, than CYP3A4 alone. This also indicates that the OATP plays major role in disposition of repaglinide. This implies that inhibition of CYP enzyme
![]()
![]() (a) (b)
(a) (b)
Figure 1. (a) Repaglinide Km estimation in MLM; (b) Repaglinide Km estimation in HLM, the concentration ranging from 0.334 µM to 243 µM for both MLM and HLM.
![]()
![]() (a) (b)
(a) (b)
Figure 2. (a) Pharmacokinetic profile of repaglinide in mice (n = 3) at 0.1 mg/kg and 0.02 mg/kg. Repaglinide dosed at 0.005 mg/kg was not detected when dosed alone; (b) Repaglinide 0.005 mg/kg dosed after a single dose RTV and KTZ one hour prior dosing of repaglinide. Doses of RTV and KTZ were 45 and 40 mg/kg respectively (n = 3). KTZ significantly increased the Cmax and AUC of repaglinide with (P < 0.001 & P < 0.05) respectively.
(3A4 and 2C8, either of them or both) along with OATP would have higher change in pharmacokinetics of repaglinide [9] . In the current study the Cmax (P < 0.001) and AUCall (p < 0.05) was significantly increased by KTZ when compared to repaglinide dosed alone.
Similarly, RTV (45 mg/kg) is a prototype irreversible CYP3A4 inhibitor and OATP inhibitor, increased repaglinide (0.005 mg/kg) concentration when dosed with RTV, the Cmax increased from below detection levels to 2.88 ng/ml and also able to calculate AUC (5.91 hr*ng/ml). Although the significance not shown at lower dose of repaglinide (0.005 mg/kg), we have to be aware that when tested at 0.1 and 0.02 mg/kg mice were moribund or dead (the significance not obtained due to higher standard deviation). In comparison with KTZ and clarithromycin (which showed clinical DDIs with repaglinide) which are CYP3A4 reversible and irreversible inhibitors respectively, RTV and AMP, both are potent irreversible inhibitor than clarithromycin and additionally RTV is also potent and non specific inhibitor the OATP and AMP is moderate inhibitor of OATP1B1 [4] [10] [11] , the possibility of RTV or AMP DDIs with repaglinide are high.
5. Conclusion
In conclusion the current result demonstrated high possibility of DDIs when RTV or AMP dosed with repaglinide due to strong DDIs in liver microsomes furthermore RTV showed DDIs in in vivo mouse model. Based on the current results future clinical trials can be planned with lower dose of repaglinide with RTV and AMP with close monitoring of patients.
NOTES
*Corresponding author.