Solvent- and Catalyst-Free Microwave-Assisted Decarboxylation of Malonic Acid Derivatives ()
1. Introduction
The synthesis of carboxylic acid derivatives via malonic ester alkylation, hydrolysis, and subsequent decarboxylation is a well-established method for carbon- carbon bond formation [1] [2] [3] , and it is one of the most important reactions in organic synthesis. Each of these steps is under constant revision with the aim of developing more efficient procedures in terms of stereocontrol, use of friendly reaction conditions and better yields [4] .
The key step in this multistep reaction sequence is the decarboxylation process, which is usually performed under harsh thermal conditions (up to 200˚C) using conventional [5] [6] or microwave heating [1] .
In thermal conditions, the decarboxylation step can be carried out using xylene, aniline, dimethylaniline, quinoline, dioxane, pyridine [7] , toluidine, xylidine [8] , catechol, resorcinol [9] and acid media [10] [11] as solvents at high temperature for long periods of time (overnight). The heating time has been reduced to an hour with the use of cumene as a solvent in the presence of catalytic amounts of pyridine [12] . In addition to those reactions a milder copper-catalyzed decarboboxylation of malonic acid derivatives was reported to proceed in acetronitrile at reflux temperature [3] .
Previous reports have established the convenience of microwave assistance as an alternative to high temperatures. For example, Giguere and collaborators carried out the decarboxylation of malonic acid derivatives such as hexanoic acid, butanoic acid, etc. in water and MW conditions: 800 W, 190˚C for 15 min [13] . Also, Helavi and colleagues have used microwave irradiation (medium to low power) to carry out the decarboxylation of malonic acid derivatives using poly- 4-vinyl pyridine as a catalyst in DMF for a short time (3 - 4 min) [14] . On the other hand, Tellitu and coworkers established a simple solvent-free protocol to promote the decarboxylation step from mono- and di-substituted malonate monoacid derivatives with short periods of irradiation (1 - 4 min) but using stoichiometric quantities of imidazole [4] .
The decarboxylation reaction has been extensively studied and milder conditions have been found. For example, malonic acid derivatives undergo decarboxylation when treated with N,N’-carbonyldiimidazole (CDI) at room temperature to generate the carbonyl imidazole moiety in high yield [1] .
In our research group, we have tried to reproduce some of the reported methods to get impurities related to Valproic acid VPA, which is used as an antiepileptic drug and a mood stabilizer [15] - [20] . But even when those methodologies allowed us to get the compounds that we were interested in, they presented drawbacks such as high temperatures for long reaction times, use of catalysts, use of difficult-to-remove solvents, elaborated workup procedures and low yields.
In this work, we report the solvent- and catalyst-free microwave-assisted decarboxylation of malonic acid derivatives; the decarboxylation process takes around 10 min at 200 W, 180˚C - 190˚C in an open vessel system. As an additional advantage, this procedure does not require any workup since the required carboxylic acid is the only product obtained.
2. Result and Discussion
Scheme 1 shows the malonic acid derivatives prepared from the commercial available diethyl 2-propylmalonate 1. To synthesize the desired compounds first the malonate 1 was converted into the corresponding enolate using NaHMDS or t-BuOK in dry THF and alkylated with the desired alkyl halides to obtain the dialkylated malonates 2a-2e in moderate to excellent yields. Then, the subsequent saponification of dialkylated malonates with potassium hydroxide in ethanol- water mixture [12] resulted in the formation of the malonic acids 3, 3a-3e with moderate to excellent yields.
Table 1 shows the results obtained from the decarboxylation of the compound 3e carried out as an effort to find the best conditions to achieve the decarboxylation
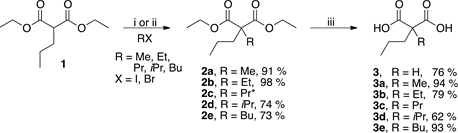
Scheme 1. Synthesis of malonic acid derivatives. Reagents and conditions: i) t-BuOK/ THF, RX, rt; ii) NaHMDS/THF, RX, −15˚C to rt; iii) KOH, H2O/EtOH, reflux [12] . * Commercially available.
![]()
Table 1. Optimization for the decarboxylation of malonic acid 3e.
Each experiment was carried out with 1.48 mmol of 3e. Experiment 3 was scaled up to 4.2 mmol yielding 60% of 4e. Experiment 4 was scaled up to 40 mmol and the yield was also quantitative.
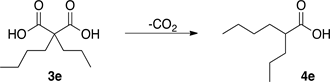
Scheme 2. Decarboxylation of malonic acid 3e.
of malonic acid derivatives, Scheme 2. Firstly, a common procedure was tested: 3e was dissolved in xylene and heated to reflux for 12 h yielding just 11% of 4e (entry 1). In a second experiment, 3e was dissolved again in xylene but this time a catalytic amount of pyridine was added [12] ; the mixture was heated to reflux for 2 h giving 37% of 4e.
In the chemical synthesis, heat is required mostly to accelerate the reaction rate and is commonly supplied by conduction through warm plate recipients. Microwave (MW) radiation is a different and very efficient alternative to heat the reaction in solution [21] [22] . Microwave-assisted organic synthesis (MAOS) is characterized by the remarkable accelerations produced in many reactions as a consequence or the heating rate, which cannot be reproduced by traditional heating. Higher yields, milder reaction conditions and shorter reaction times can be used and many processes can be improved. Indeed, even reactions that do not occur by conventional heating can be performed using microwaves [23] [24] . Therefore, Microwave (MW) activation as a non-conventional energy source that has become a very popular and useful technology in organic chemistry [25] [26] . From experiment 2, it was evident that the presence of a catalytic amount of pyridine was helpful for the decarboxylation by conventional heating and because MAOS has shown a lot of advantages over it. We decided to carry out the same reaction but under microwave irradiation (entry 3). In the third experiment, 3e was dissolved in xylene, a catalytic amount of pyridine was added and the mixture was irradiated at 200 W and 180˚C - 190˚C for 1.5 h isolating 4e with 80% yield. Scaling up experiment 3 was attempted but unfortunately the yield went down.
Microwave assisted chemistry has emerged as a discipline that permeates all aspects of synthetic chemistry. The major goals of this endeavor are to maximize the efficient use of safer raw materials and to reduce waste. On the other hand, Green Chemistry utilizes a set of principles that reduce or eliminate the use or regeneration of hazardous substances in the design, manufacture, and applications of chemical products. One of the key aspects of Green Chemistry is the elimination of solvents in chemical processes or the replacement of hazardous solvents with relatively benign solvents. The development of solvent-free alternative processes is, of course, the best solution [27] [28] [29] [30] . In this field, the microwave heating is proving to be a valuable transformation technique in sustainable chemistry as several methods have been developed using it. Some examples include the use of less solvent compared to the traditional methods and even solvent-free conditions [31] . For this reason, a fourth experiment was carried out in a microwave reactor without solvent and in the absence of catalyst (entry 4); 3e was irradiated at 200 W and heated up to 180˚C - 190˚C and it was found that the reaction finished in just 10 min. The resulting raw material did not need any treatment because 4e was completely pure; the obtained yield was excellent, 98%.
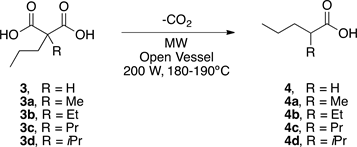
After the success of experiment 4, the decarboxylation of malonic acid derivatives 3, 3a-3d was examined under the same conditions. Table 2 summarizes the obtained results.
As we can see in all the experiments the malonic acids derivatives 3, 3a-3d were successfully decarboxylated giving the corresponding carboxylic acid 4, 4a-4d with good to excellent yields (82% - 97%) with very short reaction times (3 - 10 min). It is important to mention that because the method does not require the use of solvent and catalyst, nor does it need any workup: the products were obtained in a pure manner and no further purification was necessary.
The results show the generality, efficiency, importance and ease of this new method. In this sense, it is necessary to highlight the clearest example of what has just been mentioned and it is the quantitative yield of the decarboxylation process of 3c to get 2-propylpentanoic acid 4c (Valproic acid) in just 3 minutes. The decarboxylation step to get VPA usually requires strong bases and high temperatures for at least 2 hours [32] . On the other hand, other groups have described the synthesis VPA in short periods of time using microwave irradiation but in the presence of heterogeneous catalyst and solvents with high boiling point such as DMF [14] .
![]()
Table 2. Solvent- and catalyst-free microwave-assisted decarboxylation of malonic acid derivatives.
3. Experimental
3.1. General Information
The starting materials were obtained from commercial suppliers and were used without purification. The reactions were monitored by thin layer chromatography, on aluminum plates coated with silica gel with fluorescent indicator (60 F254). Microwave irradiation experiments were carried out in Discover CEM equipment. Melting points were measured in open capillary tubes using a Melt-temp Electrothermal apparatus, and are uncorrected. HRMS measurements were obtained on MStation JMS-700 JEOL apparatus. NMR spectra were taken with a Bruker at 300 MHz (1H) and 75 MHz (13C) and Varian Oxford at 400 MHz (1H) and 100 MHz (13C) using CDCl3 or CD3OD as solvent with TMS as internal standard.
3.2. General Procedure for the Synthesis of Dialkylmalonates 2a-2e (i)
1.2 Equiv of t-BuOK were suspended in dry THF under nitrogen atmosphere and stirred vigorously at 0˚C. After that malonate 1 (1 equiv) was added dropwise and the reaction was stirred for 20 min, then 1.2 equiv of the appropriate alkyl halide was added slowly and the reaction mixture was stirred at 0˚C for 1 h. Next, the reaction was stirred at room temperature until the malonate 1 disappeared (TLC, hexane:AcOEt 9:1). Upon completion the THF was evaporated under reduced pressure and the residue was suspended in a concentrated solution of Na2S2O3, the product was extracted three times with DCM. The combined organic fractions were dried with anhydrous Na2SO4 and concentrated. Purification of the raw oily substance by flash column chromatography (hexane:AcOEt, 98:2, 95:5, 90:10) produced the corresponding dialkylmalonate.
3.3. General Procedure for the Synthesis of Dialkylmalonates 2a-2e (ii)
1 Equiv of malonate 1 was dissolved in anhydrous THF under nitrogen atmosphere and the obtained solution was cooled to −15˚C, followed by dropwise addition of 1.1 equiv of NaHMDS (solution in THF 1M). The reaction mixture was stirred for 20 min. Then, 1.2 equiv of the appropriate alkyl halide was added slowly and the reaction mixture was stirred at −15˚C for 1 h. Next, the reaction was stirred at room temperature until malonate 1 was consumed (TLC, hexane:AcOEt 9:1). Upon completion, THF was evaporated under reduced pressure and the residue was suspended in a solution of Na2S2O3, the product was extracted three times with DCM. The combined organic fractions were dried with anhydrous Na2SO4 and concentrated. Purification of the raw oily substance by flash column chromatography (hexane:AcOEt, 98:2, 95:5, 90:10) produced the corresponding dialkylmalonate.
Diethyl 2-methyl-2-propylmalonate 2a
Colorless oil. 1H NMR (CDCl3, 300 MHz): δ (ppm) 0.89 (t, J = 7.3 Hz, 3H, CH3), 1.21 (t, J = 7.1 Hz, 6H, 2CH3), 1.17 - 1.27 (m, 2H, CH2), 1.36 (s, 3H, CH3), 1.77 - 1.82 (m, 2H, CH2), 4.14 (q, J = 7.1 Hz, 4H, 2CH2). 13C NMR (CDCl3, 75 MHz): δ (ppm) 14.2, 14.6, 17.8, 20.0, 37.8, 53.9, 61.2, 77.2, 172.7. HRMS: Calcd for C11H20O4; 216.1362; found: [M + H]+ C11H21O4, 217.1497.
Diethyl 2-ethyl-2-propylmalonate 2b
Colorless oil. 1H NMR (CDCl3, 300 MHz): δ (ppm) 0.81 (t, J = 7.6 Hz, 3H, CH3), 0.92 (t, J = 7.2 Hz, 3H, CH3), 1.11-1.19 (m, 2H, CH2), 1.24 (t, J = 7.2 Hz, 6H, 2CH3), 1.81 - 1.87 (m, 2H, CH2), 1.92 (q, J = 7.6 Hz, 2H, CH2), 4.17 (q, J = 7.1 Hz, 4H, 2CH2). 13C NMR (CDCl3, 75 MHz): δ (ppm) 8.4, 14.1, 14.4, 17.3, 25.2, 33.9, 57.9, 60.9, 171.9. HRMS: Calcd for C12H22O4; 230.1518; found: [M + H]+ C12H23O4, 231.1582.
Diethyl 2-isopropyl-2-propylmalonate 2d
Colorless oil. 1H NMR (CDCl3, 300 MHz): δ (ppm) 0.91 (t, J = 7.3 Hz, 3H, CH3), 0.97 (d, J = 6.9 Hz, 6H, CH3), 1.16 - 1.27 (m, 8H, CH2, 2CH3), 1.82 - 1.87 (m, 4H, 2CH2), 4.17 (sep, J = 6.9 Hz, 1H, CH), 4.18 (q, J = 7.1 Hz, 4H, 2CH2). 13C NMR (CD3OD, 75 MHz): δ (ppm) 14.4, 14.7, 18.2, 18.8, 32.0, 36.2, 60.8, 61.9, 171.5.
Diethyl 2-butyl-2-propylmalonate 2e
Colorless oil. 1H NMR (CDCl3, 400 MHz): δ (ppm) 0.87 - 0.94 (m, 6H, CH3), 1.24 - 1.36 (m, 12H, 3CH2, 2CH3), 1.82 - 1.89 (m, 4H, 2CH2), 4.17 (q, J = 7.1 Hz, 4H, 2CH2). 13C NMR (CDCl3, 100 MHz): δ (ppm) 14.0, 14.4, 14.6, 17.5, 23.1, 26.3, 32.0, 34.5, 57.7, 61.0, 172.2. HRMS: Calcd for C14H26O4; 258.1831; found: [M + H]+ C14H26O4, 259.1893.
2-Propylmalonic acid 3
White solid; mp 88˚C - 90˚C. 1H NMR (CD3OD, 500 MHz): δ (ppm) 0.79 (t, J = 7.4 Hz, 3H, CH3), 1.22 (sex, J = 7.5 Hz, 2H, CH2), 1.66 (q, J = 7.7 Hz, 2H, CH2), 3.14 (t, J = 7.5 Hz, 1H, CH). 13C NMR (CD3OD, 125 MHz): δ (ppm) 14.2, 21.8, 32.3, 52.9, 173.4. HRMS: Calcd for C6H10O4; 146.0579; found: [M + H]+ C6H11O4, 147.0727.
2-Methyl-2-propylmalonic acid 3a
White solid; mp 93˚C - 96˚C. 1H NMR (CD3OD, 400 MHz): δ (ppm) 0.93 (t, J=7.4 Hz, 3H, CH3), 1.24 - 1.34 (m, 2H, CH2), 1.36 (s, 3H, CH3), 1.77 - 1.81 (m, 2H, CH2). 13C NMR (CD3OD, 100 MHz): δ (ppm) 14.9, 18.9, 20.7, 54.8, 176.3. HRMS: Calcd for C7H12O4; 160.0736; found: [M + H]+ C7H12O4, 161.0811.
2-Ethyl-2-propylmalonic acid 3b
White solid; mp 90˚C - 93˚C. 1H NMR (CD3OD, 400 MHz): δ (ppm) 0.84 (t, J = 7.6 Hz, 3H, CH3), 0.93 (t, J = 7.2 Hz, 3H, CH3), 1.17 - 1.27 (m, 2H, CH2), 1.79 - 1.83 (m, 2H, CH2), 1.89 (q, J = 7.5 Hz, 2H, CH2). 13C NMR (CD3OD, 100 MHz): δ (ppm) 9.1, 14.9, 18.7, 26.9, 35.8, 59.2, 175.9. HRMS: Calcd for C8H14O4; 174.0892; found: [M + H]+ C8H14O4, 175.0968.
2,2-Dipropylmalonic acid 3c
White solid; mp 152˚C - 154˚C. 1H NMR (CD3OD, 400 MHz): δ (ppm) 0.93 (t, J = 7.2 Hz, 6H, 2CH3), 1.17 - 1.27 (m, 4H, 2CH2), 1.79 - 1.84 (m, 4H, 2CH2). 13C NMR (CD3OD, 100 MHz): δ (ppm) 14.9, 18.8, 36.4, 58.7, 175.9. HRMS: Calcd for C9H16O4; 188.1049; found: [M + H]+ C9H16O4, 189.1122.
2-Isopropyl-2-propylmalonic acid 3d
White solid; mp 119˚C - 121˚C. 1H NMR (CD3OD, 400 MHz): δ (ppm) 0.92 (t, J = 7.2 Hz, 3H, CH3), 1.00 (d, 6H, 2CH3), 1.23 - 1.33 (m, 2H, CH2) 1.81 - 1.86 (m, 2H, CH2), 2.62 (sep, J = 6.9 Hz, 1H, CH). 13C NMR (CD3OD, 100 MHz): δ (ppm) 14.9, 19.0, 19.6, 34.1, 37.7, 62.9, 175.7. HRMS: Calcd for C9H16O4; 188.1049; found: [M + H]+ C9H17O4, 189.1134.
2-Butyl-2-propylmalonic acid 3e
White solid; mp 147˚C - 149˚C. 1H NMR (CD3OD, 400 MHz): δ (ppm) 0.91 (q, J = 7.1 Hz, 6H, 2CH3), 1.14-1.25 (m, 4H, 2CH2), 1.32 (sex, J = 7.3 Hz, 2H, CH2), 1.79 - 1.86 (m, 2H, CH2). 13C NMR (CD3OD, 100 MHz): δ (ppm) 14.4, 14.9, 18.9, 24.2, 27.8, 34.1, 36.7, 58.7, 176.2. HRMS: Calcd for C10H18O4; 202.1205; found: [M + H]+ C10H19O4, 203.1294.
3.4. General Procedure for the Solvent-Free and Catalyst-Free Microwave-Assisted Decarboxylation of Malonic Acid Derivatives 3, 3a-3e
The corresponding malonic acid derivative was placed in a round-bottomed flask and was irradiated in open system (open vessel) at 200 W and 180˚C - 190˚C for 3 - 10 min in a CEM Discover microwave reactor. The reaction was monitored by TLC (hexane:AcOEt, 70:30). Further purification was not necessary because the corresponding product was pure.
Pentanoic acid 4 [33] .
Light yellow oil. 1H NMR (CDCl3, 200 MHz): δ (ppm) 0.90 (t, J = 7.2 Hz, 3H, CH3), 1.35 (sex, J = 7.3 Hz, 2H, CH2), 1.61 (quint, J = 7.4 Hz, 2H, CH2), 2.34 (t, J = 7.4 Hz, 2H, CH2), 11.09 (bs, 1H, OH). 13C NMR (CDCl3, 50 MHz): δ (ppm) 13.9, 22.4, 26.9, 34.0, 180.9.
2-Methylpentanoic acid 4a [34] . Light yellow oil. The 1H and 13C NMR are in agreement with those reported in the literature.
2-Ethylpentanoic acid 4b
Light yellow oil. 1H NMR (CDCl3, 300 MHz): δ (ppm) 0.89-0.96 (m, 6H, 2CH3), 1.25 - 1.79 (m, 6H, 3CH2), 2.25 - 2.36 (m, 1H, CH), 11.5 (bs, 1H, OH). 13C NMR (CDCl3, 100 MHz): δ (ppm) 11.9, 14.2, 20.8, 25.4, 34.1, 47.1 183.5. HRMS: Calcd for C7H14O2; 130.0994; found: [M + H]+ C7H15O2, 131.1073.
2-Propylpentanoic acid 4c [35] .
Light yellow oil. 1H NMR (CDCl3, 200 MHz): δ (ppm) 0.90 (t, J = 7.0 Hz, 6H, 2CH3), 1.21 - 1.71 (m, 8H, 4CH2), 2.30 - 2.41 (m, 1H, CH), 11.4 (bs, 1H, OH). 13C NMR (CDCl3, 50 MHz): δ (ppm) 14.2, 20.8, 34.6, 45.4, 183.7.
2-Isopropylpentanoic acid 4d
Light yellow oil. 1H NMR (CDCl3, 400 MHz): δ (ppm) 0.91 (t, J = 7.2 Hz, 3H, CH3), 0.95 (dd, J = 6.8 Hz, J = 1.2 Hz, 6H, 2CH3), 1.20 - 1.63 (m, 4H, 2CH2), 1.87 (oct, J = 6.8 Hz, 1H, CH), 2.10 - 2.15 (m, 1H, CH), 11.6 (bs, 1H, OH). 13C NMR (CDCl3, 100 MHz): δ (ppm) 14.2, 20.3, 20.7, 21.2, 30.7, 31.7, 52.7, 182.8. HRMS: Calcd for C8H16O2; 144.115; found: [M + H]+ C8H16O2, 145.1225.
2-Propylhexanoic acid 4e
Light yellow oil. 1H NMR (CDCl3, 400 MHz): δ (ppm) 0.89 (t, J = 7.3 Hz, 6H, 2CH3), 1.25 - 1.37 (m, 6H, 3CH2), 1.38 - 1.48 (m, 2H, CH2), 1.55 - 1.64 (m, 2H, CH2), 2.30 - 2.37 (m, 1H, CH), 11.4 (bs, 1H, OH). 13C NMR (CDCl3, 100 MHz): δ (ppm) 13.9, 14.0, 20.8, 22.8, 29.7, 32.1, 34.5, 45.6, 183.6. HRMS: Calcd for C9H18O2; 158.1307; found: [M + H]+ C9H18O2, 159.1377.
4. Conclusions
In conclusion, the method has shown that the decarboxylation reaction successfully proceeds with brief microwave irradiation in the absence of solvent and without catalyst. The reaction occurs with all of the synthetized malonic acid derivatives in near quantitative yield and no further workup is necessary. These conditions make this process environmentally friendly and hence very attractive for Green Chemistry.
On the other hand, since decarboxylation of malonic acid derivatives is one of the most common reactions in organic synthesis, particularly in a multistep reaction sequence this new and simple method could be useful for preparing a broad variety of biologically active compounds such as α- [36] and β-amino acids [37] , known drugs such as (S)-Dapoxetine [38] [39] one of the more effective and safe drugs for treating premature ejaculation [40] [41] [42] and Tirofiban [43] [44] an antiplatelet drug (antithrombotic).
In particular, our research group is working on microwave-assisted enantioselective decarboxylation.
Acknowledgements
We would like to thank CONACyT for financial support (Project No. CB2015/ 256653). We are also grateful to SIGNA Civac S. A de C. V. for providing the starting compounds.