1. Introduction
Biofilms, such as dental plaque, the slime that forms on surfaces in areas with water, and the biofilms found on catheter infections, are structured communities of bacteria. Biofilms are formed by the majority of bacteria in natural and pathogenic ecosystems. Unlike a simple aggregation of bacterial cells, the bacteria in a biofilm are surrounded by a mixture of polymers, known as the extracellular polymeric substance (EPS). In order to prevent the formation of harmful biofilms, it is essential to quantify bacterial growth and biofilm development as well as be able to distinguish between bacterial strains that can form biofilms from strains that cannot. However, exactly because biofilm formation is contingent on the secretion of the polysaccharides that form the EPS matrix, rates of cell division alone are not enough to quantify biofilm development.
In order to study and quantify biofilm growth, Sinorhizobium meliloti is chosen as the model organism, but other bacteria (e.g. Pseudomonas) have also been used in the literature. S. meliloti bacteria have a symbiotic relationship with alfalfa plants as they form nodules on the roots of the plants and fixate nitrogen in exchange for carbohydrates. The production of the extracellular polysaccharides (EPS) of the bacteria is essential for the establishment of this symbiosis. S. meliloti produce two types of EPS: EPS I (succinoglycan) and EPS II (galactoglucan), where low molecular weight EPS II is found to play an important role in biofilm formation [1] . Therefore, different S. meliloti strains, wild types and mutant strains that contain mutations that affect their ability to produce EPS, particularly EPS II, will vary in their ability to form a biofilm.
Research suggests that biofilm features can be quantified by optical methods [2] and via the analysis of biofilm images. For the latter, several algorithms and parameters have been developed and defined to quantify biofilms. Parametrized features of biofilms include morphological parameters such as fractal dimension and quantitative parameters such as bio-volume [3] [4] . Furthermore, there are several techniques that can be used in order to assess the mechanical properties and permeability of the EPS matrix [5] . Two such methods are Single Particle Tracking (SPT) or Multiparticle Tracking (MPT) where one or more beads are observed, respectively. Both methods are based on the beads being inside the EPS matrix of the biofilm. Due to thermal fluctuations, the beads execute Brownian motion, which is the erratic random movement of microscopic particles in a fluid, as a result of continuous bombardment from molecules of the surrounding medium. The motion of the beads is imaged via video microscopy, the images are processed via particle tracking algorithms and, finally, the particle trajectories are obtained. Using these trajectories the EPS matrix can be studied without disturbing the system [5] . In one such study, SPT was used to obtain the diffusion coefficients of differently charged beads showing in this way that surface functionalization of the particles affects their mobility in 1-day old B. multivorans and P. aeruginosa biofilms and in cystic fibrosis sputum [6] . In another study, SPT was used to find the beads’ mean-square displacement in E. coli biofilms at their 2nd and 4th day of growth so as to determine how the motion of the beads is affected by their size and charge [7] .
In other studies, rather than obtaining the diffusion coefficients associated with the Brownian motion of microspheres, the diffusion coefficients of macromolecular solutes were determined instead. This was done for various biofilm- forming bacteria, such as Pseudomonas fluorescens [8] , Staphylococcus epidermis [9] , Streptococcus mutans [10] , as well as oral biofilms [11] . Such macromolecular solutes are fluorescently tagged so that they can be easily observed and distinguished from the surrounding bacteria. In these studies the diffusion coefficient was measured by observing the fluorescent intensity of a biofilm cell cluster over time as the fluorescent macromolecules penetrated the cluster. The techniques used for this purpose were confocal laser microscopy or fluorescence recovery after photobleaching (FRAP).
In this paper, we are also using SPT to obtain the diffusion coefficients of microspheres inside S. meliloti colonies. However, this study includes mutants of the bacteria in order to determine whether the diffusion coefficient can distinguish between the strains and quantify bacterial growth. Furthermore, the strains are set to grow inside the channels of a microfluidic device. Microfluidic devices are used extensively for experimental studies in microbiology in general [12] and in biofilm formation [13] and biofilm properties [14] [15] in particular. Their low fabrication cost, small dimensions, physical properties, and ability to conduct multiple experiments on a single device under identical conditions that can be easily controlled and automated render them ideal for such studies. Because of their transparency, the use of these devices for studies in bacterial biofilms is often combined with an optical technique, such as fluorescence and confocal microscopy [14] [15] [16] [17] . Thus, in this study, the use of microfluidic devices provides a controlled environment where all S. meliloti strains grow under the same conditions and where the Brownian motion of the beads can be easily observed under the microscope. Furthermore, a microfluidic platform allows for this method to be scalable so that multiple bacterial strains can be studied simultaneously. Finally, when combined with motorized microscope stage technology, microfluidics allow for an easy automation of the data acquisition and analysis process.
1.1. Biofilms
In order to determine the viability of the diffusion coefficient as an indicator of biofilm growth, the five strains of S. meliloti used in this study include two wild types (Rm1021 and Rm8530 expR+) and three mutant strains (Rm8530 exoY, Rm9034 expG, and Rm9030-2 expA1). These strains present variation in their EPS production and synthesis via gene expression that is well documented. It has been found that the Rm1021 strain does not show extensive biofilm formation as it does not produce the low molecular weight EPS II. This is because Rm1021 carries an insertion mutation within the expR+ gene and this insertion prevents EPS II production under standard culture conditions [18] . Strain Rm8530 expR+, on the other hand, has a functional expR gene, which results in EPS II production and thereby, the formation of a highly structured biofilm [1] . The exoY gene affects the biosynthesis of EPS I [19] . Therefore, the Rm8530 exoY mutant is incapable of producing EPS I but, since it can still produce EPS II, it can form highly structured biofilms [20] . The expG is a transcriptional activator of the exp gene that regulates EPS II biosynthesis [21] . The Rm9034 expG strain has an intact functional expR gene but the expG gene has been deleted. This deletion has been found to have no effect on the level of exp gene expression in strains similar to those with the intact expR+ gene [22] . Thus, Rm9034 expG strains are able to produce EPS II and form biofilms. Lastly, the Rm9030-2 expA1 strain is missing the structural gene expA1. As a result, the EPS II polysaccharides cannot be synthesized and, thus, this strain cannot form biofilms [18] . Table 1 summarizes these characteristics.
Since these strains differ in their ability to produce EPS II and, thus, form biofilms, it is expected that beads placed in these bacterial communities will present differences in their Brownian motion: the strains with the most structured biofilms will have the most dense colonies and viscous EPS matrix, resulting in the lowest diffusion coefficients for the beads. Thus the diffusion coefficient can be an effective quantifying parameter for distinguishing between the strains and for biofilm growth.
1.2. Diffusion Coefficient
The diffusion coefficients are measured by tracking the microspheres as they randomly move within the bacterial colonies. Based on a particle’s positions along the xy plane the effective diffusion coefficient can be determined using the mean-square displacement formula
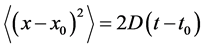 , (1)
, (1)
where x (m) is the x-coordinate of the position of the bead at time t (s), x0 is its initial x-coordinate at time t0, and D (m2/s) is the effective diffusion coefficient. The equation is similar for the bead’s motion along the y axis.
The diffusion coefficient D can be calculated theoretically using the Einstein equation for free Brownian motion
 , (2)
, (2)
where 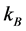 is Boltzmann’s constant (
is Boltzmann’s constant (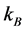 = 1.38 × 10−23 m2・kg/(s2・K)), T is the temperature (K),
= 1.38 × 10−23 m2・kg/(s2・K)), T is the temperature (K),  is the fluid viscosity (Pa・s), and
is the fluid viscosity (Pa・s), and 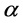 is the bead’s radius (m).
is the bead’s radius (m).
Equation (2) cannot be applied for beads moving inside a biofilm because their Brownian motion is not free. This is because D will be affected by the density of the cells within the biofilm community, the nature of the biofilm (i.e. via electrostatic interactions between the microspheres and the biofilm’s EPS matrix), as well as other physical properties (e.g., biofilm heterogeneity), all of which will be changing as the bacterial community develops. Equation (2) can, however, be used for estimating D in control experiments where the beads are seen to diffuse freely in media where there are no bacteria.
Therefore, in the work described in this paper we are examining whether the
![]()
Table 1. Strains of S. meliloti studied for biofilm formation and growth. Three out of the five strains are known to produce biofilms. Therefore, the use of the diffusion coefficient as a means of quantifying biofilm formation and development should corroborate these findings.
diffusion coefficient of green fluorescent polystyrene microspheres (0.29 μm in diameter), which execute Brownian motion in the same microfluidic channel where the bacteria are growing and potentially forming biofilms, can distinguish between strains and corroborate the results reported in the literature and summarized in Table 1 about which strains form biofilms. In addition, we are examining whether the diffusion coefficient can be used as a quantitative indicator of the colony’s growth. After injecting a mixture of bacteria-beads for each bacterial strain into the microfluidic channel, the particles’ motion is observed every 24 hours using fluorescence microscopy and particle tracking algorithms as the bacteria grow for 4 days. By tracking the particles and determining their positions, the diffusion coefficient can be calculated via Equation (1). Statistical analysis can then be applied to see if the five bacterial strains present differences in the D values of their beads - both across the strains and across time for the same strain that are statistically significant, thus demonstrating that D can distinguish between strains and can be used as an indicator of biofilm development. By also obtaining brightfield images over the 4-day period, the morphology of the bacterial colonies can complement our quantitative findings, since the biofilm-forming strains will be seen to develop structured biofilms.
If the diffusion coefficient can indeed distinguish between the strains, then this parameter, which is obtained via the cost-effective and automated technique of SPT and microfluidics, can be employed instead of a genetic analysis in order to identify an unknown strain. In addition, if the individual D values can identify the stage of the biofilm development, then the appropriate drugs can be administered for a more effective, targeted, and efficient treatment.
2. Materials and Methods
The experimental methodology was developed as summarized in Figure 1. The time interval between any two consecutive boxes 1 - 4 can be arbitrary. However, when the strains and control samples are injected in the chip (box 5) the data acquisition (box 6) needs to be performed periodically, which in this study was every 24 hours. The major components of the experimental technique are the preparation of the biofilm strains, the manufacturing of the microfluidic devices,
![]()
Figure 1. The experimental procedure and microfluidic device. Each channel (100 μm wide × 12 μm tall × ~2 cm long) of the microfluidic device (upper right corner) was filled with a strain of the bacteria-bead solution. A separate channel contained the control solution of only media and beads (not depicted). The diffusion coefficient for the microspheres was calculated via MATLAB using particle tracking algorithms and Equation (1).
and the data and statistical analysis.
2.1. Bacterial Strains
The S. meliloti strains used in this study are listed in Table 1. The strains were grown at 30˚C on Luria broth (LB) agar plates [23] for 72 hours (Figure 1, box 1). Single colonies were picked for inoculating overnight cultures of liquid LB with bacteria at 30˚C on a rotary shaker at 200 rpm (Figure 1, box 2). Streptomycin to a final concentration of 500 μg/ml was added to both the agar plates and the liquid cultures.
To ensure that all strains had the same initial cell concentration at the start of the experiment (Figure 1, box 3), a 5 μl sample was taken from each culture and placed between a glass slide and cover slip. Using brightfield microscopy with a 40× objective, the average number of cells in the field of view at 5 different regions of the glass slide was determined. Based on the average cell count for each strain, the cultures were then diluted to the same concentration (30,000 cells/μl). Green fluorescent polystyrene particles 0.29 μm in diameter (Duke Scientific, Cat. No. G300) were added to each of the diluted samples at a final bead concentration of 1:106 solids (Figure 1, box 4). In the control sample, which contained no bacteria, the beads were diluted directly in LB-Streptomycin 500 μg/ml solution to the same final concentration. The bacteria-bead solutions and the control solution were then injected into 6 channels of a microfluidic device, one for each solution (Figure 1, box 5). This injection marks the beginning of the experiment (Day 0).
2.2. Microfluidic Devices
The microfluidic devices were made using the techniques of soft lithography and chip fabrication [12] and the Sylgard 184 Silicone elastomer kit (Dow Corning Corporation) (Figure 1, box 1). A 5:1 (w/w) mixture of polydimethylsiloxane (PDMS) and curing agent was poured on the silicon wafer (mold), after the mold was treated with trichloromethylsilane (TMCS). The channels on the mold were patterned using SU-8 photoresist. The 5:1 mixture on the mold was degassed to remove air bubbles and then cured at 80˚C for 30 minutes. The PDMS was subsequently removed from the mold and cut into the chips (Figure 1, upper right). The ports of the channels were punched using M919 probe needles. The chips were then rinsed using isopropanol and acetone and dried. Each chip was placed on a clean No. 1 glass cover slip. To establish bonds between the cover slip and PDMS, the chips were cured overnight at 80˚C.
Each chip had 6 channels, one for each sample (Figure 1, upper right). The channels had approximate dimensions 100 μm (width) by 12 μm (height). Because of the design of the chip, the channels had slightly different lengths, but each channel was approximately 2 cm long. The control sample and each bacteria-beads sample were injected on Day 0 into a channel by applying pressure at 4 psi. The microfluidic device was set on the stage of an Olympus IX51 inverted fluorescence microscope. The pressure was continuously applied as the bacterial strains were left to grow for a period of 4 days.
2.3. Data and Statistical Analysis
Starting on Day 0 and every 24 hours the small polystyrene beads were observed (40× objective, FITC filter) as they executed Brownian motion within their bacterial strain at two different Regions 1 and 2 of their microfluidic channel, as shown in Figure 1, upper right. Bead aggregates did form during the 4-day period, however such aggregates were not included in our data analysis. Since the beads were already mixed with the bacteria at the beginning of the experiment, observations of the particles’ motion over the 4-day period did not disturb the bacterial colonies. The fluorescent beads were easily distinguishable from the bacterial cells under fluorescence microscopy.
Each day, snapshots of the microspheres were taken every 0.2 s for a total of 10 s (i.e., 51 images) using a Hamamatsu digital CCD camera (C8484) (Figure 1, box 6). Acquiring data for more than 10 s was not feasible since the beads would usually drift out of focus. Then the beads’ positions were tracked using a computerized centroid tracking algorithm [24] in MATLAB (Figure 1, box 7). The beads selected to be tracked were those that had not adhered to the PDMS surface of the microfluidic chip and those that were within cell clusters or very near clusters (i.e., within 5 - 10 bead diameters). Each microsphere’s diffusion coefficient for each axis (Dx and Dy) was then determined from the x and y positions and times using Equation (1). The overall effective diffusion coefficient for each bead was found by averaging Dx and Dy. Stationary beads still have a non-zero D value because of noise associated with video microscopy. In order to account for this static error, we observed 50 beads that adhered to the PDMS walls of the channels and found Dstatic = 0.01 μm2/s. This static error has been accounted for in the D values found for the moving beads [25] .
For each of the five strains and the control, 5 - 8 independent experiments were conducted. Outliers in the value of D for each strain (or control) and day were subsequently removed using a cutoff of 3 standard deviations from the mean. The data points (after the removal of outliers) for each day and sample ranged from 39 to 92. The averages and standard deviations for each sample and day were then calculated. Since the number of acceptable data points was greater than 30, parametric statistical tests were performed (two-tail two-sample mean z-tests for large samples) in order to determine whether differences in the D values across days and strains were statistically significant. A total of 105 such z-tests were conducted, as explained in Section 3. In particular, it was expected that the D values associated with the control, which contained no bacteria, would show no changes over time. Furthermore, for Day 0, since there was such a low initial concentration of bacteria in the bacterial samples, it was expected that the D values corresponding to the strains and the control would not be statistically different. An ANOVA test with α = 0.01 confirmed these expectations. In addition, the D values for each day of the control were compared to each other via z-tests (10 tests). Also, z-tests were used to compare the values of D from Day 0 for each of the 5 strains to Day 0 of the control and to each other (15 tests). Since it was determined that there was no statistically significant difference between all these D values, these 25 tests constituted the checkpoints. The smallest p-value of these checkpoint z-tests (0.0036) was taken as the α value (level of significance) for all the remaining z-tests which were conducted across days and strains. The remaining z-tests included: 1) for Days 1 - 4, each strain was compared to the corresponding control for that day (20 tests), 2) for each strain, tests were conducted to see if changes in D between consecutive days were significant (20 tests), and 3) 40 tests were conducted in order to compare the strains to each other across different days.
3. Results
The observed values of the diffusion coefficients for all strains and days are shown in Figure 2. It can be seen that the D value remains the same (no statistically significant differences) across all days for the control. Furthermore, D is the same for all strains on Day 0 and is the same as the value for the control, as was
![]()
Figure 2. The diffusion coefficients over time of the microspheres in the five S. meliloti strains. The error bars represent one standard deviation (±σ) from the mean. Each bar corresponds to the average of at least 39 D values across at least 5 and at most 8 experiments.
confirmed by statistical analysis. These results were expected, since the data taken on Day 0 represent the bacteria-bead solution when it was just inserted in the microfluidic channel. Therefore, the beads are not affected by the presence of the bacteria at this low initial bacterial concentration. As mentioned in Section 2 these results correspond to the checkpoints and gave the value α = 0.0036 that was used as the level of significance for the remaining z-tests. For Days 1 - 4 all D values from the strains were significantly different from the corresponding control value.
Figure 2 also shows that, as time progresses, the diffusion coefficient decreases, with the one exception of strain Rm8530 expR+ where D increases from Day 3 to Day 4. The amount by which D decreases, however, is not the same across strains and days. To investigate whether D changes significantly between two adjacent days for each strain, two-sample mean z-tests for large samples were conducted. Table 2 summarizes the results of these parametric tests.
In Table 2, a down arrow ($) represents a statistically significant decrease in D between the two consecutive days, an up arrow (#) represents an increase in D that was statistically significant, and a dash (−) indicates no statistically significant change in D. It is clear that each strain exhibits a different behavior of changes in D over time. In addition, strains Rm8530 expR+, Rm8530 exoY, and Rm9034 expG, the biofilm-forming strains, show a statistically significant decrease in D from Day 1 to Day 2. For Rm8530 expR+ and Rm8530 exoY, D decreases by more than a factor of 10, but the decrease is not as large for Rm9034 expG. For the non-biofilm forming strains Rm1021 and Rm9030-2 expA1, on the other hand, there was no change from Day 1 to Day 2. Furthermore, as mentioned, Rm8530 expR+ was the only strain for which there was an increase in D
![]()
Table 2. Summary of the results of the two-sample z-tests for consecutive days of the five S. meliloti strains. The up arrows (#) and down arrows ($) correspond to statistically significant increases and decreases in D, respectively, while dashes (−) indicate no statistically significant changes. Each strain shows a unique pattern of up arrows, down arrows, and dashes.
between any two consecutive days. This increase occurred between Day 3 and Day 4.
In addition to performing statistical analysis between consecutive days for the same strain, two-sample mean z-tests for large samples were also conducted across strains and days. In particular, when examining the extent to which strains Rm8530 expR+, Rm8530 exoY, and Rm9034 expG are similar to each other, it was seen that Rm8530 expR+ and Rm8530 exoY have corresponding D values that are statistically different from each other only on Day 4. Furthermore, Rm9034 expG is always significantly different from both Rm8530 expR+ and Rm8530 exoY when comparing D values for the same day. However, Rm9034 expG’s value on Days 2 and 4 is not different from Rm8530 expR+ and Rm8530 exoY’s values on Days 1 and 2, respectively. This suggests that Rm9034 expG exhibits the same growth behavior in the 4-day period as Rm8530 expR+ and Rm8530 exoY in their first 2 days, but at a slower pace.
The quantitative description of the bacterial growth via the diffusion coefficient was supplemented by a qualitative description that was conducted based on the morphology (i.e., appearance and cell motility) of the bacterial colonies under brightfield microscopy. Table 3 summarizes the morphology of the strains for each day of growth and Figure 3 shows a representative picture for each state. The states that were observed were planktonic, large clusters, small clusters, biofilm, swarming, cell aggregation (with moving cells), dense cell aggregation (with moving cells), stationary cell aggregation, or some combination of these states. For strains Rm1021 and Rm9030-2 expA1 the cell aggregations of Day 4 were so dense that they likely included many dead bacteria. Figure 3 shows that the states are distinguishable. Furthermore, the EPS II-producing strains were seen to develop structured biofilms and, thus, the images provided a visual confirmation of the biofilm-forming strains [1] .
4. Discussion
Careful examination of the results associated with the control samples was the first step in ensuring the validity of the experimental methodology. Indeed, the average value of the diffusion coefficient in the control experiments across all
![]()
Table 3. Summary of the morphological states of the strains on each day of growth. The abbreviations stand for Planktonic State (PS), Large Clusters (LC), Small Clusters (SC), Biofilm (B), Swarming (S), Cell Aggregation with moving cells (CA), Dense Cell Aggregation with moving cells (DCA), and Stationary Cell Aggregation (SCA). Combinations of states were also observed.
![]()
Figure 3. Representative pictures of the bacterial growth stages. The stages are distinguishable and show the differences in morphology. The bright spots in the pictures are the fluorescent beads. Bead aggregates were observed (as seen, for example, in the picture for Rm9034 expG, Day 2), but such aggregates were not tracked. Beads stuck on the walls of the channels were analyzed in order to account for the static error in particle tracking (Section 2.3). All channels are 100 μm wide and the scale bars are 25 μm.
days is D = 1.84 ± 0.53 µm2/s and is in agreement with the values reported in the literature for diffusion coefficients in aqueous environments when accounting for the radius of the beads and the temperature of the solutions [2] [10] . Similarly, when using the viscosity of water η = 0.89 mPa・s at T = 25˚C in Equation (2), the theoretical value obtained is D = 1.69 µm2/s. This value falls within the experimental range, although the viscosity of the LB-Streptomycin mixture is different from that of water and the temperature inside the channels where the beads move is not exactly 25˚C. The latter is especially true during the image acquisition process, since the sample is illuminated by light of high intensity. Nevertheless, the experimental value for D given by the controls is confirmed by Equation (2).
Figure 2 shows that the set of D values for each strain is unique. Based on their sets of D values, the biofilm-forming strains Rm8530 expR+, Rm8530 exoY, and Rm9034 expG seem to develop in a similar manner. As mentioned, Rm8530 expR+ and Rm8530 exoY had corresponding D values that were different only on Day 4, which can be accounted for by the fact that their biofilms exhibited different behavior on that day (Table 3).
Also, Rm9034 expG seems to develop a biofilm in the same way as Rm8530 expR+ and Rm8530 exoY but at approximately half the rate, as was confirmed by the statistical analysis. Thus, it seems that Rm8530 exoY’s inability to produce EPS I does not affect the rate of biofilm development, though it seems that this strain does not reach the swarming stage as fast as the wild type Rm8530 expR+. Furthermore, the results suggest that the missing expG gene from strain Rm9034 expG does affect the biofilm’s rate of development, implying that having both genes expR+ and expG leads to faster production of EPS II and, thus, a greater biofilm growth rate. Taking data at shorter time intervals (<24 hours) can further determine the amount by which Rm9034 expG develops slower than Rm8530 expR+ and Rm8530 exoY.
As shown in Table 2, the pattern of the changes in D is unique for each strain. This observation suggests that D can be used as an identifier of each strain. Also, the statistically significant decrease in D from Day 1 to Day 2 (Table 2) that was exhibited by only the biofilm-forming strains suggests that this decrease observed in the time interval Day 1 - 2 is an important indicator of whether a strain will form a biofilm. Thus, D can be used as an indicator of biofilm formation.
As expected, no statistically significant changes were found: 1) across days for the control and 2) across the strains and control for Day 0 (checkpoint results). However, for Days 1 - 4 the five strains had a D value that was significantly different from that of the control sample. This indicates that the growth of the bacteria and the biofilm formation have a considerable effect on the diffusion coefficients of the microspheres. It is true, though, that different strains presented similar D values even when the strains were not at the same morphological state. For example, strain Rm8530 exoY on Day 1, when the bacteria were in a state of large clusters and planktonic cells, showed a D value that was comparable to that of strain Rm8530 expG on Day 2, when the bacteria were in small clusters. Similar D values but different morphologies were observed not only across strains and days but also for the same strain (e.g., no change in D for Rm8530 expG between Day 2 (small clusters) and Day 3 (large clusters)).
Similarly, different strains can show like changes in their respective values of D on consecutive days; however that does not imply that the strains will show the same morphological changes. For example, all three biofilm-forming strains, Rm8530 expR+, Rm8530 exoY, and Rm9034 expG, showed a decrease in D from Day 1 to Day 2. However, when the microfluidic channels of these strains were observed under the brightfield microscope, the morphological changes were different: Rm8530 expR+ and Rm8530 exoY clearly showed a transition from a state where the cells had formed large-area clusters or were in a planktonic state on Day 1 to a biofilm on Day 2. Strain Rm9034 expG, on the other hand, transitioned from a planktonic state on Day 1 to a state where the cells were in small clusters on Day 2 (Table 3).
Given these observations, the diffusion coefficient can therefore be used to quantify biofilm formation for biofilm-forming strains. By observing changes in D for a particular strain, the strain can be identified via Table 2. Then, knowing the strain, individual D values can be used to identify the stage of biofilm formation via Figure 2 and Table 3. However, for Rm9034 expG, where there was no significant difference between the D values for Days 2 and 3, D cannot distinguish between the states of small vs. large clusters. Therefore, it seems that the size of the clusters does not affect the diffusion of the microspheres in biofilm- forming strains. Given that we were only looking at beads that were within cell clusters (Section 2.3) this result is not surprising. Nevertheless, it does suggest that the structure and composition of the clusters does not change with cluster size for this strain.
An analogous observation can be made for distinguishing between a densely populated planktonic state, a cell aggregation, and a dense cell aggregation, as is shown for strain Rm9030-2 expA1 where the D values are not significantly different for Days 1 - 3. Again this implies that for this strain, since D decreases only slightly from Day 1 to Day 3, other than an increase in cell density, nothing else happens to the structure and composition of the bacterial colony that significantly affects the motion of the microspheres. Conversely, from Day 0 to Day 1, strains Rm9034 expG, Rm1021, and Rm9030-2 expA1 remained in the same morphological state (planktonic) and yet showed a significant change in their respective D values between these two days. This suggests that changes in D cannot be due to morphological changes alone. Therefore, D in this case is sensitive enough to distinguish between states that are morphologically very similar. Thus, changes in D might have been caused by environmental changes (e.g., changes in viscosity due to EPS production, etc.) that were not apparent when the bacterial colonies were observed under the microscope. Using quantitative parameters (e.g., fractal dimension, bio-volume [4] ) to describe the morphology of the bacterial community will facilitate the numerical correlation between its diffusion coefficient and its morphological state. Furthermore, allowing the experiment to continue for a time period that is longer than 4 days and acquiring data at time intervals that are smaller than 24 hours will allow for additional growth stages to be detected.
As the cell cultures transition from state to state (Table 3), it is seen that D can change (Table 2). Potential reasons why D changes include:
1) EPS production that increases the environment’s viscosity,
2) electrostatic interactions between the beads and the EPS matrix [7] ,
3) the irregular structure of the EPS matrix [26] ,
4) interactions between bacteria and beads, especially when the bacteria are at a high concentration, as is the case in cell clusters,
5) high density of stationary cells that effectively trap the beads,
6) motion of cells (e.g. swarming) that forces the microspheres’ motion,
or a combination of the above. Based on Table 3 and Figure 3, it is likely that each strain presents a unique set of combinations of the above reasons as to why the diffusion coefficient changes from day to day, which explains why each strain shows a unique pattern of changes in D, as presented in Table 2.
5. Conclusion
Based on the above observations, this study shows that the diffusion coefficient D of beads that are executing Brownian motion inside bacterial colonies can be used as an identifier of biofilm-forming strains and as a quantitative parameter of the stage of biofilm development. A strain can be identified via its unique set of changes in the diffusion coefficient over the 4-day period of observation. Also, the biofilm-forming strains show a statistically significant decrease in D from Day 1 to Day 2. Furthermore, there was a clear correlation between the value of the diffusion coefficient and the morphological state of the bacterial colony. Introducing the beads with the bacteria in the microfluidic channels allows for the measurements to be made using video microscopy and particle tracking algorithms without disturbing the bacterial colonies. Furthermore, using microfluidics as a platform for this experimental study provides a controlled environment for the simultaneous growth of the five bacterial strains, scalability, and automation. Thus, the combination of SPT, microfluidics, and the use of the diffusion coefficient as the parameter of interest provide an inexpensive and non-invasive alternative to genetic analysis for strain identification and characterization. This, in turn, can result in timely and targeted treatments. Though S. meliloti was the organism used in this study, the same technique can be easily applied to simultaneously study multiple strains and mutations of other biofilm-forming bacteria and, thus, the applications can be extended to include other harmful bacterial strains.
Authors’ Contribution
C. C., Y. D., M. D. and F. K. contributed equally to this work.
Acknowledgements
We would like to thank Nitya P. Jacob, Max Teplitski, and Mengsheng Gao for not only providing us with the bacteria strains used in this experiment, but also for giving us information that helped us interpret our results. E.S. is also grateful to Daniel T. Gillespie for his feedback on the manuscript. We would also like to thank the members of the Division of Natural Science and Mathematics at Oxford College for providing us with the resources needed to conduct our experiments. Funding was provided by the Howard Hughes Medical Institute under Grant No.52005873, by the Oxford College of Emory University, and by the Pierce Institute for Leadership and Community Engagement at Oxford College. Any opinions, findings, and conclusions or recommendations expressed in this work are those of the authors and do not necessarily reflect the views of the Howard Hughes Medical Institute, the Pierce Institute, or Emory University.