Molecular Modeling and Synthesis of New Heterocyclic Compounds Containing Pyrazole as Anticancer Drugs ()
1. Introduction
Microtubule-targeting agents are used clinically as antitumor drugs. Their anticancer activity is commonly due to tubulin inhibition. They prevent the formation of microtubule, thus affecting cellular replication and shape [1] . In general, antitubulin agents bind with five totally different binding pockets on the β-tubulin subunit [2] . Agents that strongly react with colchicine binding site induce depolymerization of tubulin, therefore they inhibit tubulin action [3] (Figure 1(a)). This study will focus on agents that target colchicine-binding site [4] .
The natural product CA-4 that is under investigation as it has a strong affinity to colchicine binding site, and therefore a strong tubulin inhibitor. Thus, we use
![]()
Figure 1. (a) Tubulin inhibitors that bind to colchicine binding site; (b) Common structure characteristic of CA-4.
CA-4 as a lead molecule for synthesis of new derivatives and more studied as tubulin inhibitors [5] . It is formed of three parts (2 carbons bridge, ring A and ring B) [6] (Figure 1(b)). Various changes to CA-4 structure have been performed in literature. The changes to the central bridge affect the cytotoxic activity the most [7] - [15] . Ring A has received little or no attention while ring B has received larger attention. Impressed by these finding, the double bond of CA-4 is substituted by α, β-unsaturated ketone, pyrazole, quinoline-3-carbonitriles, fused pyrimidine, fused quinazoline moieties, imine derivatives and 2-amino-4H-chromenes. Besides, ring A and B are replaced by 1, 5-diphenylpyrazole, naphthalene and pyrazole moieties. Those were evaluated in vitro for their anticancer tendency toward four cancer cell types. Additionally, molecular docking studies are performed and compared to biological results.
2. Materials and Methods
2.1. Chemistry
The new compounds are prepared by different pathways described in Scheme 1 and Scheme 2. The starting compound 3 was obtained via Vilsmeir Haack’s formylation of the pyrazole derivatives using POCl3/DMF [16] . The α, β unsaturated carbonyl derivatives 4a, 4b were synthesized via Claisen-Schmidt condensation [17] of cyclic ketones 4a, b with substituted pyrazole-4-carbaldehyde 3 in an ethanolic solution of sodium hydroxide [18] [19] [20] [21] [22] . Cyclocondensation of enone 4a with hydrazine hydrate or phenyl hydrazine using ethanol or glacial ethanoic acid afforded pyrazoles 5a-c [23] .
2-Substituted quinoline-3-carbonitrile products were made by the reaction of
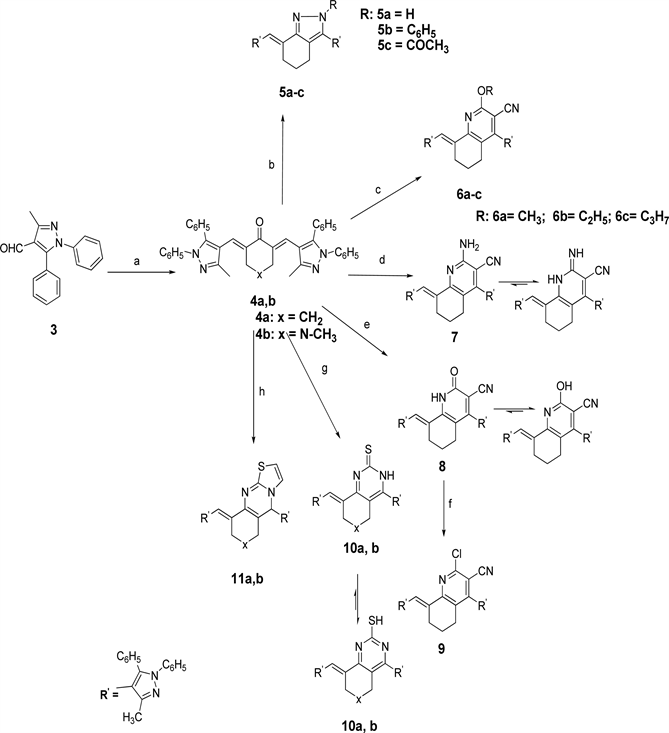
Scheme 1. Reagents and conditions: (a) cyclohexanone or N-methyl piperidone, alchoholic NaOH; (b) NH2NHR, absolute ethanol or NH2NH2, AcOH; (c) malononitrile, sodium methoxide, sodium ethoxide or sodium propoxide, methanol, ethanol or n-propanol; (d) malononitrile, ammonium acetate, absolute ethanol; (e) ethylcyanoacetate, ammonium acetate, absolute ethanol; (f) POCl3\DEA; (g) thiourea, Na metal, butanol; (h) 2-aminothiazole, AcOH.
malononitrile with compounds 4a in sodium alkoxide to give the corresponding 2-alkoxyquinoline-3-carbonitriles derivatives 6a-c. Furthermore, a reaction of chalcone 4a with malononitrile and ammonium acetate in ethanol gave 2-aminoquinoline-3-carbonitrile derivative 7. Also, 2-oxoquinoline-3-carbonitrile 8 was successfully synthesized by cyclocondensation reaction of compound 4a with ethylcyanoacetate and ammonium acetate in absolute ethanol. In another hand, chloro product 9 was collected by the reaction of compound 8 with POCl3 in catalytic amount of N, N-diethylaniline (DEA). Quinazoline-2-thione 10a, pyrido[4,3-d]pyrimidine-2-thione analogue 10b were obtained by reaction of chalcone 4a,b with thiourea in refluxing NaOBu in butyl alcohol [21] . Moreover, new derivatives of thiazolo[2,3-b]quinazoline 11a and thiazolo[3,2-a]pyrimidine 11b were prepared by reacting 2-aminothiazole with the enones 4a,b in glacial ehanoic acid [18] .
Other targeted imino derivatives 12a, b and 13 were prepared via condensation of the pyrazole carbaldehyde 3 with the appropriate aromatic amine in glacial ethanoic acid [24] [25] (Scheme 2). Additionally, substituted malononitrile
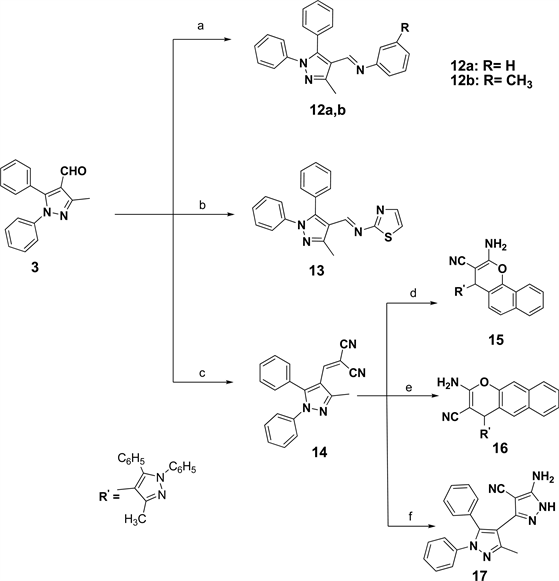
Scheme 2. Reagents and conditions: (a) aniline or m-toluidine, AcOH; (b) 2-aminothiazole, AcOH; (c) malononitrile, absolute ethanol; (d) α-naphthol, methansulfonic acid; (e) β-naphthol, methansulfonic acid; (f) NH2NH2, absolute ethanol.
nol. 2-Amino-4H-chromenes 15 and 16 were prepared from condensation of 14 was prepared from condensation of compound 3 with malononitrile in ethamalononitrile derivative 14 with naphthols using methanesulfonic acid [26] . Also, it was found that 5-amino-4-cyanopyrazole analogue 17 resulted when two moles of malononitrile derivative 14 condensed with one mole of hydrazine hydrate in absolute ethanol. All the analytical and spectroscopic data of the prepared compounds were fully accordance with their represented structures.
2.2. Biology
Four neoplastic cell types namely; (MCF-7), (HCT-116), (Hela) and (PC3) are obtained through VACSERA, Cairo, Egypt. Combretastatin A-4 was used as reference for judgment on anticancer activity of newly synthesized molecules.
The reagents RPMI-1640 medium, MTT, DMSO and CA-4 (sigma co., St. Louis, USA), Fetal Bovine blood serum (GIBCO, UK) [27] .
The MTT assay was applied to assess cytotoxic activity using the cell lines mentioned above. This type of colorimetrical chemical assay relies on the modification of the yellow tetrazolium bromide (MTT) to a color of formazan derivative by mitochondrial succinate dehydrogenase in viable cells. Cell lines were cultured in RPMI-1640 medium with ten percent fetal bovine blood serum. Then, antibiotics added were one-hundred units/ml penicillin and one-hundred µg/ml streptomycin at 37˚C in a five percent CO2 incubator. Then, drugs were prepared in different concentrations and added to the cells, incubated for 24 hrs. After the period was completed, MTT solution (20 µl) with concentration 5 mg/ml was added to the mixture and incubated for 4 hrs. Then, DMSO (100 µl) is sided to every single plate to remove purple color of formazan. The colorimetrical chemical assay is measured and recorded at an absorbance of 570 nm employing a plate reader (EXL 800, USA) [28] [29] .
2.3. Molecular Modeling
The docking studies and modeling calculations were done using MOE version 2008.10 release of Chemical Computing Groups using Windows XP software. The 3D structures of the synthesized molecules were built using Chem Biooffice suite then saved as Mol2 format with the help of MOE software, then subjected to docking simulation. The three dimensional X-ray structure of protein (PDB code: 1SA1) was obtained from the Protein Data Bank.
3. Results
3.1. Antitumor Activity
All the synthesized compounds were evaluated for their cytotoxic activity toward a panel of 4 human neoplastic cell lines using the MTT assay. The results, in Table 1 showed that every compound have antitumor activities with noticeable differences due to structural variations.
As shown in Table 1, compound 5b with 2-phenyl substituted indazole
![]()
Table 1. Cytotoxic activity (IC50, μg/ml) of (4-17).
IC50 = IC50 (μg/ml): 1 - 10 (very strong). 11 - 20 (strong). 21 - 50 (moderate). 51 - 100 (weak) and above 100 (non-cytotoxic). CA-4 = combretastatin A-4.
shows wide spectrum cytotoxic activity with IC50 starting from 6.9 to 12.7 μM. Replacement of phenyl ring with acetyl group as in compound 5c or unsubstituted indazole in compound 5a and uncyclized chalcone as in compound 4a exhibited a decrease within the activity against the four cell lines. Additionally, the cytotoxic activities of recent synthesized quinoline-3-carbonitrile 6-9, compounds 6a with 2-methoxy and 6c with 2-propoxy group has broad spectrum activity virtually identical IC50, (22.6 ± 1.87 for HCT-116, 14.9 ± 1.23 for Hela, 17.7 ± 1.41 for PC3, 14.0 ± 1.41 for MCF-7), (19.8 ± 1.50 for HCT-116, 16.0 ± 1.32 for Hela, 18.9 ± 1.39 for PC3, 13.8 ± 1.42 for MCF-7) respectively. In comparison to compound 6c, replacing propoxy group with ethoxy group in compound 6b resulting in 1.5 times decrease in cytotoxic activity. Also replacing propoxy group with amino group in compound 7 resulting in reduce in activity by almost 4 times. Compounds 8 and 9 shows low activity within the four cell lines. By replacing CH2 of cyclohexanone chalcone 4a by N-CH3 chalcone 4b, the cytotoxic activity is almost doubled against the four human cell lines. While the cytotoxic activity of 10a and 11b is ranged from moderate to weak activity but compounds 10b and 11a are inactive with IC50 > 100 μg/ml.
Also, the series of 4-substituted 1, 5-diphenyl pyrazole shows a range of intermediate to high cytotoxicity activity. Compounds with benzo chromene-3- carbonitrile substitution as in 15 and 16 results in very strong cytotoxic activity with IC50, (10.8 ± 0.92 for HCT-116, 9.5 ± 0.84 for Hela, 13.7 ± 0.97 for PC3, 8.8 ± 0.93 for MCF-7) and (9.0 ± 0.68 for HCT-116, 7.8 ± 0.42 for Hela, 7.9 ± 0.68 for PC3, 12.2 ± 0.81 for MCF-7) respectively. Replacement of aniline or substituted aniline in compounds 12a, b by either 2-amino thiazole in compound 13 or substituted pyrazole in compound 17 enhance cytotoxic activity with IC50, (11.4 ± 0.98 for HCT-116, 14.4 ± 1.05 for Hela, 15.6 ± 1.20 for PC3, 9.1 ± 1.10 for MCF-7) and (17.4 ± 1.32 for HCT-116, 13.1 ± 1.10 for Hela, 16.3 ± 1.36 for PC3, 10.7 ± 1.23 for MCF-7) respectively.
3.2. Molecular Modeling
Molecular docking was performed for compounds that inhibited tubulin polymerization so as to explore attainable binding modes. The tubulin protein structure in complex with podophyllotoxin (PDB 1SA1) was used for the study [30] [31] [32] . In molecular docking, MOE programme (molecular operating environment) was used in our study. CA-4 is employed as lead compound due to its excellent affinity to colchicine binding site (Figure 2). Comparing to the lead molecule CA-4 and biological activity, we have a tendency to found that compounds 5b, 15, 16 and 17 have the lowest energy and give the most stable complexes with the receptor and are the most biological active against cell lines so we concluded that these new compounds have very good affinity binding with the protein. During this study, we observed that basic amino acid Lys (B254) is essential as it is bound with 15 via arene cation interaction and with 16 via hydrogen bond (angle NH…N = 101.9, distance = 2.9 A0) (Figure 4, Figure 5). Also, it was found that Tyr (A224) is important for arene-arene interaction with 5b, 15 and 17 (Figure 3, Figure 4, Figure 6). Also, Glu (A183) is bound to the amino group in 15 (Figure 4) via hydrogen bond (angle NH…O = 111.9, distance = 2.8 A0). A Hydrogen bond is formed between a nitrogen atom in nitrile group in 17 (Figure 6) and hydroxyl side chain of ser (A140) (angle N…HO =1 02.9, distance = 2.7 A0).
4. Experimental
Melting points (˚C) were recorded using a Fisher-Johns melting point apparatus and are uncorrected. Microanalyses (C, H, and N) were performed in Cairo University Microanalytical unit, and all the results within ±0.4. IR spectra (KBr) were recorded on Thermo Fisher Scientific Nicolet IS10. Spectrometer (ν in
![]()
Figure 2. Binding of CA-4 with 1SA1 amino acids. (a) Gln: Glutamine; (b) Lys: Lysine; (c) Ala: Alanine; (d) Ser: Serine.
![]()
Figure 3. Binding of 5b with 1SA1 amino acids. (a) Tyr: Tyrosine.
![]()
Figure 4. Binding of 15 with 1SA1 amino acids. (a) Lys: Lysine; (b) Tyr: Tyrosine; (c) Glu: Glutamic acid; (d) Mg+2 cation.
![]()
Figure 5. Binding of 16 with 1SA1 amino acids. (a) Lys: Lysine.
![]()
Figure 6. Binding of 17 with 1SA1 amino acids. (a) Tyr: Tyrosine; (b) Ser: Serine.
cm−1) at Faculty of Pharmacy, Mansoura University. Mass MS (EI) m/z analyses were performed on Thermo Scientific DCQII, Faculty of Science, Mansoura University. 1H and 13C-NMR spectra were obtained in DMSO-d6 or CDCl3 on ASCEND spectrometer (400 MHz) Bruker at Georgia State University USA, Faculty of Pharmacy, Zagazig University and Faculty of Science, Kafr ElSheikh University. Reaction times were decided using TLC on Silica gel plates 60 F254 (E. Merk), and the spots were visualized by U.V (366, 245 nm). Compounds 2, 3 were prepared as previously discussed in literature [16] [33] .
4.1. General Method for Preparation of Chalcones 4a, b
Ethanolic solution (20 ml) of NaOH (1 g, 0.025 mol) was mixed with a solution of 3-methyl-1,5-diphenyl-1H- pyrazole-4-carbaldehyde (3) (1 g, 0.02 mol) and the appropriate cyclic ketones (0.01 mol) in ethyl alcohol (30 ml). The previous mixture was stirred at 25˚C for 2 h. The product obtained was then filtered, washed with water, dried and finally recrystallized from ethyl alcohol.
(2E,6E)-2,6-bis[(3-Methyl-1,5-diphenyl-1H-pyrazol-4-yl)methylene]cyclohexanone (4a) as yellow solid; yield 45% (1 g); mp 230˚C - 233˚C; 1H NMR (DMSO-d6): δ 7.2 - 7.3 (m, 20H, Ar-H), 7.1 (d, J = 8.1 Hz, 2H, methylene-H), 2.5 (s, 6H, 2CH3), 2.1 (s, 4H, cyclohexanone-H), 1.6 (q, J = 7.1 Hz, 2H, cyclohexanone-H); 13C NMR: δ 13 (2c), 25.7 (1c), 26.1 (2c) 115.7 (2c), 124.5 (4c), 126.2 (2c), 127.5 (4c), 128 (2c), 129.2 (4c), 129.3 (4c), 133 (2c), 139.8 (2c), 142.2 (2c), 143.8 (2c), 147.9 (2c), 150 (2c), 191 (1c); IR (KBr, cm-1): 1670 (C=O), 1627 (C=N), 1547, 1489 (C=C); MS (EI) m/z: 586 (30%, M+); anal (calcd.) for C40H34N4,C: 81.88, H: 5.84, N: 9.55. Found, C: 81.87, H: 5.85, N: 9.04.
(3E,5E)-1-Methyl-3,5-bis[(3-methyl-1,5-diphenyl-1H-pyrazol-4-yl)methylene]piperidin-4-one (4b) as brown solid; yield 75% (0.8 g); mp 300˚C - 302˚C; 1H NMR (DMSO-d6): 7.1 - 7.7 (m, 22H, Ar-H, 2H-methylene), 4.1 (s, 4H, CH2NCH2), 2.5 (s, 6H, 2CH3), 2.2 (s, 3H, N-CH3); IR (KBr, cm-1): 1670 (C=O); MS (EI) m/z: 601 (98.9%, M+); anal (calcd.) for C40H35N5,C: 79.84, H: 5.86, N: 11.64. Found, C: 79.88, H: 5.85, N: 11.55.
4.2. General Procedure for Synthesis of 5a, c
Hydrazine hydrate (98%) (0.3 ml, 0.05 mol) introduced to a solution of 4a in ethyl alcohol (99%) (20 ml) 5a or ethanoic acid (10 ml) 5c and the mixture was refluxed for 5 h. The solvent was decreased using evaporator. After cooling the separated product was filtered, dried and finally recrystallized from ethyl alcohol.
4.3. General Procedure for Synthesis of 5b
A mixture of chalcone 4a (0.7 g, 0.005 mol), phenyl hydrazine (0.12 g, 0.005 mol) in glacial ethanoic acid (15 ml) was heated for 8 h. Then the mixture was diluted with ice-cold water. The precipitate was collected, washed with water, dried and purified by SiO2 (preparative TLC in petroleum ether: ethyl acetate 3:1)
3-(3-Methyl-1,5-diphenyl-1H-pyrazol-4-yl)-7-(E)[(3-methyl-1,5-diphenyl-1H-pyrazol-4-yl)methylene]-3,3a,4,5,6,7-hexahydro-2H-indazole (5a): (1 g, 0.01 mol) of compound 4a gave 5a as white powder; yield 40% (0.4 g); mp 150˚C - 155˚C; 1H NMR (DMSO-d6): δ 8.1 (s, 1H, NH (D2O exchangeable), 7.1 - 7.3 (m, 21H, 20 Ar-H, methylene-H), 2.5 (s, 3H, CH3), 2.2 (t, J = 7.7 Hz, 2H, indazole H-4), 2.1 (s, 3H, CH3), 1.9 (m, 4H, indazole-H5, H6); IR (KBr, cm-1): absence of C=O at 1670, 3410 (NH); MS (EI) m/z: 600 (8.8%, M+); anal (calcd.) for C40H34N6, C: 79.97, H: 6.04, N: 13.99. Found, C: 79.85, H: 6.01, N: 13.78.
3-(3-methyl-1,5-diphenyl-1H-pyrazol-4-yl)-7-(E)[(3-methyl-1,5-diphenyl-1H-pyrazol-4-yl)methylene]-2-phenyl-3,3a,4,5,6,7-hexahydro-2H-indazole (5b): as yellow solid; yield 83% (0.5 g); mp 129˚C - 130˚C. 1H NMR (DMSO-d6): δ 7.4 - 7.7 (m, 26H, Ar-H, methylene-H), 2.7 (t, J = 7.3 Hz, 2H, indazole-H4), 2.4 (s, 3H, CH3-pyrazole), 2.1 (s, 3H, CH3-pyrazole), 1.9 (m, J = 7.1 Hz, 4H, indazole -H5, H6); IR (KBr, cm-1): 1598 (C=C), absence of C=O at 1670; MS (EI) m/z: 676 (31%, M+); anal calcd for C46H38N6: C: 81.63, H: 5.96, N: 12.42. Found: C, 81.53, H: 5.87 N: 12.38.
1-{[3-(3-Methyl-1,5-diphenyl-1H-pyrazol-4-yl)-7-(E)[(3-methyl-1,5-diphenyl-1H-pyrazol-4-yl)methylene]-3,3a,4,5,6,7-hexahydro-2H-indazol-2-yl}ethanone (5c): (0.7 g, 0.005 mol) of compound 4a gave 5c as buff solid; yield 57% (0.4 g); mp 138˚C - 140˚C; 1H NMR (DMSO-d6): δ 7.1 - 7.3 (m, 21H, 20 Ar-H, methylene-H), 2.7 (t, J = 7.7 Hz, 2H, indazole H-4), 2.2 (s, 3H, COCH3), 2.5 (s, 3H, CH3-pyrazole), 2.1 (s, 3H, CH3-pyrazole), 1.9 (m, 4H, indazole-H5, H6); IR (KBr, cm-1): 1656 (C=O), 1504 (C=C); MS (EI) m/z: 642 (59%, M+); anal (calcd.) for C42H36N6, C: 78.33, H: 5.85, N: 13.99. Found, C: 78.5, H: 5.01, N: 13.78.
4.4. Preparation of 6a-c
A mixture of compound 4a (0.6 g, 1 mmol) and malononitrile (0.07 g, 1 mmol) was added to freshly prepared solution of sodium alkoxide (15 ml), (0.3 g, 0.014 mol) of sodium in 100 ml of the appropriate alcohol. The mixture kept at room temperature for the ideal time. The triggered product became collected by filtration, washed with ethanol and recrystallized from ethanol/ DMF.
2-Methoxy-4-(3-methyl-1,5-diphenyl-1H-pyrazol-4-yl)-8-(E)[(3-methyl-1,5-diphenyl-1H-pyrazol-4-yl)methylene]-5,6,7,8-tetrahydroquinoline-3-carbonitrile (6a) as white solid; yield 66% (0.4 g); m.p 160˚C - 163˚C; 1H NMR (DMSO-d6): δ 8.1 (s, 1H, methylene-H), 7.4 - 7.7 (m, 20H, 20Ar-H), 4.1 (s, 3H, OCH3), 3.1 (t, J = 7.5 Hz, 2H, quinoline-H5),, 2.5 (s, 6H, 2CH3), 2.1 (m, 4H, quinoline-H6, H7); 13C NMR: δ 11.6 (1c), 12.5 (1c), 21.5 (1c), 26.7 (1c), 32.2 (1c), 54.1 (1c), 111.7 (1c), 112.2 (1c), 116.3 (1c), 119.7 (1c), 120.8 (1c), 123.9 (2c), 124.8 (2c), 126.5 (1c), 127 (1c), 128.2 (1c), 128.4 (2c), 128.51 (4c), 128.6 (4c), 129.3 (2c), 129.8 (2c), 129.9 (2c), 130.3 (1c), 131 (1c), 139.4 (1c), 139.6 (1c), 139.9 (1c), 140.5 (1c), 141 (1c), 147.2 (1c), 147.4 (1c), 159.5 (1c); IR (KBr, cm-1): absence of CO peak at 1670, 2221 (CN), 1598 (CN ring),; MS (EI) m/z: 664.9 (69%, M+); anal. calcd for C44H36N6: C: 79.49, H: 5.46, N: 12.64. Found: C: 79.2, H: 5.41, N: 12.64.
2-Ethoxy-4-(3-methyl-1,5-diphenyl-1H-pyrazol-4-yl)-8-(E)[(3-methyl-1,5-diphenyl-1H-pyrazol-4-yl)methylene]-5,6,7,8-tetrahydroquinoline-3-carbonitrile (6b) as buff solid; yield 50% (0.3 g); mp 130˚C - 135˚C; 1H NMR (DMSO-d6): δ 6.5 - 7.3 (m, 21H, 20Ar-H, H-methylene), 3.8 (m, 2H, OCH2), 3.1 (t, 2H, J = 7.1 Hz, quinolineH-5), 2.5 (s, 6H, 2CH3), 2.1 (m, 4H, quinoline-H6, H7), 1.6 (t, J = 7.5 Hz, 3H, CH3); IR (KBr, cm-1): absence of CO, 2220 (CN); MS (EI) m/z: 678 (12%, M+); anal. calcd for C45H38N6: C: 79.62, H: 5.64, N: 12.38. Found: C: 79.21, H: 5.43, N: 12.04.
4-(3-Methyl-1,5-diphenyl-1H-pyrazol-4-yl)-8-(E)[(3-methyl-1,5-diphenyl-1H-pyrazol-4-yl)methylene]-2-propoxy-5,6,7,8-tetrahydroquinoline-3-carbonitrile (6c) as white solid; yield 71% (0.5 g); mp 141˚C -142.5˚C; 1H NMR (DMSO-d6): δ 8.1 (s, 1H, H-methylene), 7.5 -7.8 (m, 20H, 20Ar-H), 3.8 (t, J = 7.8 Hz, 2H, OCH2), 3.1 (t, 2H, J = 7.1 Hz, quinoline-H5), 2.5 (s, 6H, 2CH3), 1.9 (m, 4H, quinolineH6, H7), 1.8 (m, 2H, C-CH2), 1.6 (m, 3H,CH3); 13CNMR: δ 11.6 (2c), 12.5 (2c), 21.5 (1c), 26.7 (1c), 32.3 (1c), 54.1 (1c), 111.7 (1c), 112.2 (1c), 116.3 (1c), 119.7 (1c), 120.8 (1c), 123.9 (2c), 124.8 (2c), 126.5 (1c), 127 (1c), 128.2 (1c), 128.4 (2c), 128.51 (2c), 128.59 (2c), 128.6 (2c), 128.7 (2c), 129.3 (2c), 129.8 (2c), 129.9 (2c), 130.3 (1c), 131 (1c), 139.4 (1c), 139.6 (1c), 139.9 (1c), 140.5 (1c), 141 (1c), 147.2 (1c), 147.4 (1c), 159.5 (1c); IR (KBr, cm-1): absence of CO, 2221 (CN), 1598 (CN ring); MS (EI) m/z: 692 (20%, M+); anal. calcd for C46H40N6: C: 79.74, H: 5.82, N: 12.13. Found: C: 79.03, H: 5.31, N: 12.11.
4.5. Preparation of 2-Amino-4-(3-Methyl-1,5-Diphenyl-1H-Pyrazol-4-yl)-8-(E) [(3-Methyl-1,5-Diphenyl-1H-Pyrazol-4-yl) Methylene] -5,6,7,8-Tetrahydroquinoline-3-Carbonitrile (7)
Compound 4a (2.6 g, 0.005 mol) was mixed with malononitrile (0.3 g, 0.005 mol) and ammonium acetate (2.7 g, 0.04 mol) in ethyl alcohol (99%) (30 ml).The mixture was refluxed for 7 h, and then concentrated under reduced pressure. After cooling, the precipitated product was filtered, dried and purified by SiO2 (preparative TLC petroleum ether: ethyl acetate 3:1) to yield compound 7 as yellow powder; yield 71% (2 g); mp 300˚C - 302˚C; 1H NMR (DMSO-d6): δ 7.1 - 7.3 (m, 21H, Ar-H, H-methylene), 6.7 (s, 2H, NH2 (D2O exchangeable), 3.1 (t, 2H, J = 7.1 Hz, quinoline-H5), 2.5 (s, 3H, CH3), 2.1 (s, 3H, CH3), 1.9 (m, 4H, quinolineH6, H7); IR (KBr, cm-1): 2215 (CN), 3413 (NH2), 1598 (C=N ring); MS (EI) m/z: 649.3 (20.3%, M+); anal. calcd for C43H35N7: C: 79.33, H: 5.4, N: 15.4. Found: 79.46, H: 5.4, N: 15.09.
4.6. Preparation of 4-(3-Methyl-1,5-Diphenyl-1H-Pyrazol-4-yl)-8-(E) [(3-Methyl-1,5-Diphenyl-1H-Pyrazol-4-yl) Methylene]-2-Oxo- 1,2,5,6,7,8-Hexahydroquinoline-3-Carbonitrile (8)
Compound 4a (1.5 g, 0.01 mol) was mixed with ethylcyanoacetate (0.3 g, 0.1 mol) and ammonium acetate (2 g, 0.1 mol) in ethyl alcohol (99%) (50 ml), the reaction mixture was refluxed for 15 h. After cooling, the formed precipitate was filtered, washed with ethyl alcohol, then washed with water, dried and purified by SiO2 (preparative TLC petroleum ether: ethylacetate 3:1) to yield compound 8 as yellow powder; yield 41% (0.7 g); mp 152˚C - 153˚C. 1H NMR (DMSO-d6): δ 9.1 (s, 1H, NH (D2O exchangeable), 7.1 - 7.7 (m, 20H, Ar-H), 6.3 (s, 1H, H-methylene), 3.3 (m, 4H, quinoline-H5, H7), 2.1 (s, 6H, 2CH3), 1.9 (m, 2H, quinoline-H6); IR (KBr, cm-1): 2221 (CN), 3400 (NH); MS (EI) m/z: 650 (57%, M+); anal. calcd for C43H34N7: C: 79.48, H: 5.4, N: 12.78. Found: C: 79.33, H: 5.4, N: 12.91.
4.7. Preparation of 2-Chloro-4-(3-Methyl-1,5-Diphenyl-1H-Pyrazol-4-yl)-8-(E)[(3-Methyl-1,5-Diphenyl-1H-Pyrazol-4-yl) Methylene]- 1,2,5,6,7,8-Hexahydroquinoline-3-Carbonitrile (9)
A mixture of 8 (0.7 g, 0.0013 mol), phosphorus oxychloride (2.5 ml, 0.026 mol) and N,N-diethylaniline (DEA) (0.1 ml) was heated for 6 h. After cooling by ice (15 ml), the product turned into filteration, dried and washed with ice-water to yield compound 9 as yellow powder; yield 71% (0.5 g); mp 190˚C - 192˚C; 1H NMR (DMSO-d6): δ 7.1 - 7.7 (m, 20H, Ar-H), 6.1 (s, 1H, H-methylene), 3.3 (m, 4H, quinoline-H5, H7), 2.1 (s, 6H, 2CH3), 1.9 (m, 2H, quinoline-H6). IR (KBr, cm-1): 2220 (CN), absence of C=O band at 1670; MS (EI) m/z: 673 (11%, M++2), 671 (20%, M+); anal. calcd for C43H33ClN6: C: 76.85, H: 5.85, Cl: 5.13, N: 12.46. Found: C: 76.2, H: 5.11, Cl: 5.5, N: 12.2.
4.8. Preparation of 10a, b
The α, β-unsaturated ketone 4a or 4b (0.01 mol) was mixed with thiourea (0.8 g, 0.01 mol) and Sodium metal (0.5 g) in butyl alcohol (50 mL). The mixture was refluxed for 10 h. The extra of solvent became concentrated in vacuo and the residue changed into pH 6. The separated stable turned into filteration, washed with water, dried and purified through SiO2 (preparative TLC petroleum ether: ethyl acetate 3:1).
4-(3-Methyl-1,5-diphenyl-1H-pyrazol-4-yl)-8-(E)[(3-methyl-1,5-diphenyl-1H-pyrazol-4-yl)methylene]-4,4a,5,6,7,8-hexahydroquinazoline-2(3H)-thione (10a): As yellow solid; yield 20% (0.3 g); mp 138˚C - 140˚C; 1H NMR (DMSO-d6): δ 8.3 (s, 1H, SH), 7.1 - 7.3 (m, 20H, 20Ar-H), 5.4 (s, 1H, H-methylene), 2.8 (m, 4H, quinazoline-H5, H7), 2.4 (s, 6H, 2 CH3), 1.6 (m, 2H, quinazoline-H6); MS (EI) m/z: 644 (51%, M+); anal. calcd for C41H34N6S: C: 76.37, H: 5.63, N: 13.03, S: 4.97. Found: C: 76.2, H: 5.11, N: 13.2, S: 4.5.
6-Methyl-4-(3-methyl-1,5-diphenyl-1H-pyrazol-4-yl)-8-(E)[(3-methyl-1,5-diphenyl-1H-pyrazol-4-yl)methylene]-4,4a,5,6,7,8-hexahydropyrido[4,3-d]pyrimidine-2(3H)-thione (10b): As brown solid; yield 15% (0.2 g); mp 150˚C - 153˚C; 1H NMR (DMSO-d6): δ 9.2 (d, 1H, SH), 7.1 - 7.3 (m, 20H, 20Ar-H), 4.8 (s, 1H, H-methylene), 3.3 (s, 4H, pyridine-H2,H6), 2.4 (s, 6H, 2CH3), 2.2 (s, 3H, N-CH3); MS (EI) m/z: 659.1 (5.5%, M+); anal. calcd for C41H35N7S: C: 74.63, H: 5.65, N: 14.86; S: 4.86. Found: C: 74.2, H: 5.55, N: 14.2, S: 4.67.
4.9. Preparation of 11a, b
A solution of 2-aminothiazole (0.08 g, 0.01 mol) and the α,β-unsaturated ketone 4a or 4b (0.5 g, 0.01 mol) in glacial ethanoic acid (20 ml) was refluxed for 20 h. Solvent was dried under reduced pressure; then washed with water. The obtained product was recrystallized from ethyl alcohol.
5-(3-Methyl-1,5-diphenyl-1H-pyrazol-4-yl)-9-(E)[(3-methyl-1,5-diphenyl-1H-pyrazol-4-yl)methylene]-6,7,8,9-tetrahydro-5H-thiazolo[2,3-b]quinazoline (11a): As yellow crystals; yield 80% (0.4 g); mp 200˚C - 201˚C; 1H NMR (DMSO-d6): δ 7.1 - 7.3 (m, 22H, 20Ar-H and 2Thiazole-H), 6.3 (s, 1H, H-methylene), 4.5 (s, 1H, thiazolo quinazoline-H5), 2.8 (m, 2H, thiazolo quinazoline-H8), 2.5 (s, 6H, 2 CH3), 1.6 (m, 4H, thiazolo quinazoline-H6, H7). 13CNMR: δ 13 (1c), 14 (1c), 23.3 (1c), 23.2 (1c), 25 (1c), 56 (1c), 97 (1c), 113 (1c), 119 (1c), 124 (1c), 124.5 (4c), 126.4 (2c), 127.5 (4c), 128.8 (2c), 129.9 (8c), 132 (2c), 133 (3c), 134.1 (1c), 139.2 (2c), 142 (1c), 146 (1c), 148.7 (1c), 150 (1c), 152.9 (1c), 158.4 (1c); MS (EI) m/z: 668 (13%, M+); anal. calcd for C43H36N6S: C: 77.22, H: 5.43, N: 12.56, S: 4.79. Found: C: 77.20, H: 5.81, N: 12.40, S: 4.91.
7-Methyl-5-(3-methyl-1,5-diphenyl-1H-pyrazol-4-yl)-9-(E)[(3-methyl-1,5-diphenyl-1H-pyzol-4-yl)methylene]-6,7,8,9-tetrahydro-5H-pyrido[4,3-d]thiazolo[3,2-a]pyrimidine (11b): As brown crystals; yield 80% (0.4 g); mp 130˚C - 133˚C; 1H NMR (DMSO-d6): δ 7.1 - 7.3 (m, 22H, 20Ar-H and 2Thiazole-H), 6.3 (s, 1H, H-methylene), 4.5 (s, 1H, pyrimidine-H4), 3.1 (m, 4H, pyridine H2, H6), 2.4 (s, 6H, CH3), 2.2 (s, 3H, N-CH3); IR (KBr, cm-1): 1529 (C=N ring); MS (EI) m/z: 683.1 (5%, M+; anal. calcd for C43H37N7S: C: 75.52, H: 5.45, N: 14.34, S: 4.69. Found: C: 75.22, H: 5.82, N :14.40, S: 4.95.
4.10. Preparation of 12a, b and 13
A mixture of compound 3 (2 g, 0.1 mol) and appropriate aromatic amine in glacial ethanoic acid (20 ml) was heated under reflux for 5 h. The solid was precipitated by adding cold water. The separated solid turned into filteration, washed with water, dried and purified by means of SiO2 (preparative TLC petroleum ether: ethyl acetate 3:1).
[1-(3-Methyl-1,5-diphenyl-1H-pyrazol-4-yl)]-N-phenylmethanimine (12a): aniline (0.8 g, 0.1 mol) to yield pure white powder; yield 38% (1 g); mp 120˚C - 122˚C; 1H NMR (DMSO-d6): δ 8.1 (s, 1H, CH=N), 7.2 - 7.4 (m, 15H, Ar-H), 2.6 (s, 3H, CH3 -pyrazole); 13C NMR: δ 14.3 (1c), 116.9 (1c), 120.5 (2c), 125 (2c), 125.2 (1c), 127.6 (1c), 128.1 (1c), 128.7 (2c), 128.9 (2c), 129.1 (2c), 129.2 (2c), 130.2 (1c), 138.8 (1c), 145.7 (1c), 148.7 (1c), 152.7 (1c), 153.5 (1c); MS (EI) m/z: 337.1 (40.4%, M+); anal. calcd for C23H19N3: C: 81.87; H, 5.68; N, 12.45. Found: C: 81.45, H: 5.81, N: 12.74.
[1-(3-Methyl-1,5-diphenyl-1H-pyrazol-4-yl)-N-(3-methylphenyl)]methanimine (12b): m-toluidine (0.7 g, 0.1 mol) to yield brown powder; yield 40%(1 g); mp 120˚C - 122˚C; 1H NMR (DMSO-d6): δ 8.1(s, 1H, CH=N), 7.2 - 7.4 (m, 14H, Ar-H), 2.4 (s, 3H, CH3 -pyrazole), 2.2 (s, 3H, CH3); MS (EI) m/z: 351.1 (38.7%, M+); anal. calcd for C24H21N3: C: 82.02; H:6.02; N: 11.96. Found: C: 82.45, H: 6.81, N: 11.74.
[1-(3-Methyl-1,5-diphenyl-1H-pyrazol-4-yl)-N-(thiazol-2-yl)]methanimine (13): 2-aminothiazole (0.7 g, 0.1 mol) to yield buff powder; 62% (1.5 g); mp 130˚C - 131.5˚C; 1H NMR (DMSO-d6): δ 8.1 (s, 1H, CH=N), 7.1 - 7.8 (m, 12H, Ar-H), 2.5 (s, 3H, CH3 -pyrazole). anal. calcd for C20H16N4S: C: 69.74; H: 4.68; N: 16.27, S: 9.31. Found: C: 69.43, H: 4.81, N: 16.74, S: 9.22.
4.11. Preparation of [2-(3-Methyl-1,5-Diphenyl-1H-Pyrazol-4-yl) Methylene] Malononitrile (14)
Mixture of methyl-1, 5-diphenyl-1H-pyrazole-4-carbaldehyde 3 (3.5 g, 1 mol), malononitrile (0.9 g, 1 mol), in ethyl alcohol 95% (20 ml) were refluxed for 12 h. After of entirety of the reaction with the aid of TLC, the mixture changed into cooling to 25˚C, filtered and washed with water. The resulting solid product was recrystallized from methanol to yield compound 14 as pure yellow crystals 75% (3 g); mp 135˚C - 136˚C; 1H NMR (DMSO-d6): δ 8.1 (s, 1H, ethylenic-H), 7.4 (d, 3H, J = 5 Hz, Ar-H), 7.3 (s, 3H, Ar-H), 7.2 (d, 4H, J = 5 Hz, Ar-H), 2.5 (d, 3H, J = 5 Hz, CH3).13C NMR: δ 14.2 (1c), 80.7 (1c), 113.3 (1c), 114.2 (1c), 114.6 (1c), 125.3 (2c), 127.5 (1c), 128.5 (1c), 129 (1c), 129.1(1c), 129.8 (1c), 129.9 (2c),138.3 (2c), 146.1 (1c), 149.1 (2c), 154.3 (1c); IR (KBr, cm-1): 2221 (CN), 1597 (C=C, C=N of ring); MS (EI) m/z: 310.1 (100%, M+); anal. calcd for C20H14N4: C: 77.40; H: 4.55; N: 18.05. Found: C: 77.43, H: 4.81, N: 18.43.
4.12. Preparation of 15, 16
Mixture of compound 14 (3.5 g, 1 mmol), the appropriate naphthol (1 mmol) and methanesulfonic acid (1.2 g, 1 mmol) in acetonitrile (5 ml) were refluxed for 5 h. After completion of the reaction, the aggregate turned into cooling to room temperature and filtered. The filtrate wad washed by 5% NaHCO3 and dried over MgSO4. The solvent turned into evaporated. The resulting solid product turned into recrystallization from methyl alcohol.
[2-Amino-4-(3-methyl-1,5-diphenyl-1H-pyrazol-4-yl)]-4H-benzo[h]chromene-3-carbonitrile (15): Pure white crystals; yield 50% (3 g); mp 120˚C - 121˚C. 1H NMR (DMSO-d6): δ 7.2 - 7.4 (m, 16H, Ar-H), 6.9 (s, 2H, NH2 (D2O exchangeable), 4.7 (s, 1H, benzochromene-H4), 1.7 (s, 3H, CH3); IR (KBr, cm-1): 2188 (CN), 3448 (NH2); anal.cald for C30H22N4: C: 79.27; H: s4.88; N: 12.33. Found: C: 79.6; H: 4.5; N: 12.8.
[2-Amino-4-(3-methyl-1,5-diphenyl-1H-pyrazol-4-yl)]-4H-benzo[g]chromene-3-carbonitrile (16): To give pure white crystals; yield 40% (2 g); mp 150˚C - 151˚C. 1H NMR (DMSO-d6): δ 9.7 (s, 2H, NH2 (D2O exchangeable), 7.1 - 7.7 (m, 16H, Ar-H), 3.3 (s, 1H, H-4benzochromene), 2.5 (s, 3H, CH3); IR (KBr, cm-1): 2188 (CN), 3448 (NH2); MS (EI) m/z: 454.3 (10.5%, M+); anal. calcd for C30H22N4: C: 79.27, H: 4.88, N: 12.33. Found: C: 79.43, H: 4.81, N: 12.74.
4.13. Preparation of 17
A solution of compound 16 (4.6 g, 2 mol) in15 ml of ethanol was added to 80% hydrazine hydrate (0.6 ml, 1 mol). The mixture was stirred at 25˚C for 4 hrs, the solvent was evaporated under reduced pressure. The residue was washed with ethyl alcohol, recrystallized from the water.
[5-Amino-3’-methyl-1’,5’-diphenyl-1H,1’H-3,4’-bipyrazole]-4-carbonitrile (17): As white needles; yield 40% (1 g); mp 197˚C - 198˚C; 1H NMR (DMSO-d6): δ 7.5 (s, 1H, NH (D2O exchangeable), 7.1 - 7.4 (m, 10H, Ar-H), 6.4 (s, 2H, NH2(D2O exchangeable), 2.5 (d, 3H, J = 5 Hz, CH3); IR (KBr, cm-1): 2200 (CN), 3420 (NH2); MS (EI) m/z: 340 (13.3%, M+); anal. calcd for C20H16N6: C: 70.57, H: 4.74, N: 24.69. Found: C: 70.43, H: 4.25, N: 24.74.
5. Conclusion
A series of new compounds have been synthesized using efficient methods and tested for their biological activities. There is a strong correlation between molecular modeling and biological screening results which suppose that the structural modification of the lead structure affects the activity in a predictable manner; as shown in (Figures 2-6), CA-4 shows highest binding affinity with its methoxy phenyl moiety and 2C bridge. Compound 5b with 6C bridge and the methoxy phenyl moiety is replaced by diphenyl pyrazole ring gives good binding with colchicine binding site. Compounds 15 and 16 with a 4C spacer as double bond is replaced by chromene, ring A is replaced by diphenyl pyrazole moiety and ring B is replaced by naphthalene, have a good binding affinity with the protein but compound 15 have the best binding affinity. In compound 17, after removing 2C distance, replacing ring A and B by substituted pyrazoles, we found that the activity as tubulin inhibitor highly increased and it’s been considered as strong colchicine binding site agent.
Acknowledgements
Authors wish to thank Faculty of pharmacy, Mansoura University and Delta University for science and Technology for funding and using their laboratories equipment. Thanks for National Cancer Institute, Cairo, Egypt.