Comparison of Two Possible Evolutionary Mechanisms of the 62 tRNA Codon Sequences ()
1. Introduction
There are many reports on mitochondrial (mt) tRNA mutations would linked with inherited diseases [1] [2] [3] , i.e., mutations in tRNAIle, tRNALys and tRNASer(UCN) are known to cause different mt encephalomyopathies or non-syndromic deafness [3] . The mutation of tRNA genes has a research hotspot. Many tRNA sequences have been found and deposited in a database [4] and they all conform to one of 62 codon sequence groups (two codon groups are absent from the database, i.e., aaa and cta). All modern tRNA sequences have evolved from a common ancestor, but their evolutionary mechanism remains an open question. Two possible mechanisms have been proposed to explain the origin and evolution of modern tRNAs, i.e., “point mutation” and “template duplication” [5] . The “point mutation” mechanism suggests that divergence occurred mainly via “mutation” [6] [7] [8] [9] [10] . It is hypothesized that a new sequence may be recruited from the mutant RNA molecule or be recruited from an isoaccepting group by another point mutation in the sequence that concurrently changed the tRNA amino acid identity and its messenger RNA coupling capacity, which is known as “codon capture” or “tRNA gene recruitment” [11] . In contrast to evolution via mutation, the “template duplication” mechanism proposes that tRNAs diverged from a precursor via a duplication mechanism. It is hypothesized that primordial tRNAs with hairpin structures associate themselves with a template and produce another RNA via sequence complementarity [12] - [17] . It is thought that four initial pairs of pre-tRNAs with complementary anti-codons could have been capable of generating all 64 anticodons.
Theoretically, if modern tRNAs evolved via point mutations, the matching scores of sequences that align in a parallel direction, 5’ 3’ vs. 5’ 3’, should be larger than the scores aligned in an antiparallel direction, 5’ 3’ vs. 3’ 5’. If modern tRNAs evolve by complementary duplication method, however, the opposite would be true. To identify the most likely mechanism for tRNA evolution, we compared tRNA sequences in parallel and antiparallel directions, and we constructed two types of networks to make a comparison. The corresponding parallel and antiparallel phylogenetic trees of the 62 codon sequences could be generated by altering the parameters of these networks using the neighbor-joining method to different degrees. We discuss the evolutionary properties of the codon sequences for the 62 tRNA groups and the two types of phylogenetic trees.
2. Materials and Methods
2.1. Experimental Data
The 3695 tRNA sequences are acquired from the database (http://trnadb.bioinf.uni-leipzig.de/) [1] and the highly similar species in the database were filtered. Each tRNA sequence contained 76 bases and we removed the bases in the variable stem and the poly-A tail.
2.2. tRNA Network Construction
2.2.1. Important Network Parameters
Degree and clustering coefficient: the network is made up of a set of nodes (vertices) and the connections between them (edges or links). The features and the nature of the network were indicated by two main parameters [18] , i.e., the degree, k, and the clustering coefficient, c, of the nodes. The degree k of a node was the number of nodes with which it was connected. If a node connected with i other nodes and j edges had connections within these i nodes, the clustering coe- fficient c of the original node was defined as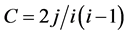 , where
, where 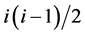 is the total number of possible connections among i nodes. The clustering coefficient reflected the relationships among the neighbors of a node and it quantified the inherent tendency of the network to cluster. Investigating the degree and clustering coefficient distribution of the network facilitated the study of the local properties among the nodes and their global statistics.
is the total number of possible connections among i nodes. The clustering coefficient reflected the relationships among the neighbors of a node and it quantified the inherent tendency of the network to cluster. Investigating the degree and clustering coefficient distribution of the network facilitated the study of the local properties among the nodes and their global statistics.
2.2.2. Method for Constructing the tRNA Network
The steps used to construct the tRNA network were as follows.
(1) Compare the two tRNA sequences stochastically, ith and jth, in parallel direction and antiparallel directions, and record their degree of similarity, 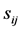 where
where 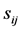 is defined as the alignment scores for the ith and jth tRNAs (shown in Figure 1)
is defined as the alignment scores for the ith and jth tRNAs (shown in Figure 1)
(2) Consider each tRNA as one node in the network and add a connection if two tRNAs have a degree of similarity, s, that is larger than a given degree of similarity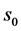 .
.
After repeating these two steps, an undirected tRNA network can be constructed based on a given degree of similarity. In this study, the 3695 tRNA sequences were divided into 62 groups based on their different tRNAs anticodons. The networks were constructed using different degrees of similarity in parallel and antiparallel directions, and the corresponding average degree 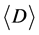 and average clustering coefficient
and average clustering coefficient 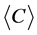 of these networks were recorded.
of these networks were recorded.
2.3. Phylogenetic Tree Construction
We constructed the tRNA phylogenetic trees suing the neighbor-joining method [19] . To determine the evolutionary relationships among the 62 tRNA codon sequences, we used a novel method to compute the evolutionary distances between different tRNAs groups using two important parameters,
and
, for different networks of tRNAs, rather than comparing only two sequences. The trees were constructed using the following steps.
(i) We used the ith and jth codon groups to produce larger tRNA groups and used them to construct a network, before calculating the average degree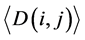 and the average clustering coefficient
and the average clustering coefficient 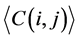 for this larger network.
for this larger network.
(ii) The evolutionary distance between any two codon groups was computed using the following equation:
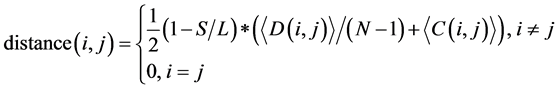 (1)
(1)
![]()
Figure 1. The degree of similarity of two tRNA segments in two directions, parallel: , antiparallel:
, antiparallel: . The alignment score was defined as the degree of similarity, i.e., the number of bases in two tRNAs that were the same in a parallel comparison, such as A-A, C-C, T-T, and G-G, or complementary in an antiparallel comparison, such as A-T, C-G, and G-T.
. The alignment score was defined as the degree of similarity, i.e., the number of bases in two tRNAs that were the same in a parallel comparison, such as A-A, C-C, T-T, and G-G, or complementary in an antiparallel comparison, such as A-T, C-G, and G-T.
where i and j ran from 1 to 62. In this study, L was the length of one tRNA, and N was the total number of nodes in the network. We use 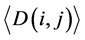 and
and 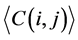 to represent the alignment scores of two different codon groups, which showed the evolutionary affiliation of tRNAs with two different codons, which helped us to interpret the overall evolutionary trend of the tRNA sequences to some extent.
to represent the alignment scores of two different codon groups, which showed the evolutionary affiliation of tRNAs with two different codons, which helped us to interpret the overall evolutionary trend of the tRNA sequences to some extent.
(iii) We constructed the phylogenetic tree for the 62 tRNA codon sequences using PHYLIP 3.67 and TreeView 1.6.6.
3. Results
Figure 2 shows the diverse relationships among the phylogenetic trees of the 62 tRNA codon groups. Each tree branch represents one group with a specific codon sequence. Figure 2(a) shows that a degree of similarity of 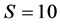 produced two large clusters, i.e., ((att,(cct,(act,(tgt,(tga,(cga,(tat,(tct,(gct,(aag:agt))))))))))), (cgg,(tca,(gca,(agg,(tcg,(atc,(acc:acg))))))), gcc,(gcg,(gtg:tgg)) and ((cgc,(gta,(gtc,(gac,(aca,(ctg,(ggg:ccc)))))))), agc,(cca,(acc,(gtt,(aga,(gga,(ctt:ggt)))))),ggc,(ctc,(tcc:ttg)), as well as 15 isolated branches and one small cluster in the overall parallel phylogenetic tree, where each large cluster comprised three small clusters. the sequences atg, aac, tta, ata, caa, ttt, cat, gat, gag, tag, gaa, and taa were found in isolated branches, but their evolutionary distance was small. They exhibited a regular ladder ranked appearance. All of the codons in the large clusters supported the point mutation mechanism and each had a matching codon with only one base difference in the same site. We also found that some codons of the isoaccepting tRNAs appeared in the same cluster and neighboring clusters. For example, six codons the amino acid Arg, i.e., cct, tct, tcg, acg, ccg, and gcg, for were in the same cluster, three of them were in a neighboring branch, and three were dispersed among different branches. In contrast to the parallel phylogenetic tree for s = 10, the antiparallel phylogenetic tree for s = 10 only contained two large clusters and some isolated branches, and the codons of these two large clusters exhibited a regular laddered rank. Thus, these results did not support “template duplication mechanism.” Some codons of isoaccepting tRNA sequences also appeared in the same cluster, e.g., four codons for Ser were in the same cluster, whereas the other two codons were in another cluster. Most codons for isoaccepting tRNA sequences were dispersed throughout the tree.
produced two large clusters, i.e., ((att,(cct,(act,(tgt,(tga,(cga,(tat,(tct,(gct,(aag:agt))))))))))), (cgg,(tca,(gca,(agg,(tcg,(atc,(acc:acg))))))), gcc,(gcg,(gtg:tgg)) and ((cgc,(gta,(gtc,(gac,(aca,(ctg,(ggg:ccc)))))))), agc,(cca,(acc,(gtt,(aga,(gga,(ctt:ggt)))))),ggc,(ctc,(tcc:ttg)), as well as 15 isolated branches and one small cluster in the overall parallel phylogenetic tree, where each large cluster comprised three small clusters. the sequences atg, aac, tta, ata, caa, ttt, cat, gat, gag, tag, gaa, and taa were found in isolated branches, but their evolutionary distance was small. They exhibited a regular ladder ranked appearance. All of the codons in the large clusters supported the point mutation mechanism and each had a matching codon with only one base difference in the same site. We also found that some codons of the isoaccepting tRNAs appeared in the same cluster and neighboring clusters. For example, six codons the amino acid Arg, i.e., cct, tct, tcg, acg, ccg, and gcg, for were in the same cluster, three of them were in a neighboring branch, and three were dispersed among different branches. In contrast to the parallel phylogenetic tree for s = 10, the antiparallel phylogenetic tree for s = 10 only contained two large clusters and some isolated branches, and the codons of these two large clusters exhibited a regular laddered rank. Thus, these results did not support “template duplication mechanism.” Some codons of isoaccepting tRNA sequences also appeared in the same cluster, e.g., four codons for Ser were in the same cluster, whereas the other two codons were in another cluster. Most codons for isoaccepting tRNA sequences were dispersed throughout the tree.
For s = 35, a comparison of Figure 2(a) and Figure 2(c) showed that the distribution of the parallel phylogenetic tree in Figure 2(c) was clearly different, with fewer isolated branches while the codons in the large cluster were more dispersed. Two small clusters and one large cluster comprised the whole tree, where most codons had matching codons with only one base difference in the small cluster, although a few had no mutated codons in the upstream branch. More codons of the isoaccepting tRNA sequences were present in the same cluster and they were closer. As shown in Figure 2(d), the distribution of the anti-
parallel phylogenetic tree with s = 35 was more compact compared with the parallel phylogenetic tree with s = 35, i.e., three small clusters, one large cluster, and two isolated branches comprised the entire tree. Two clusters formed the core of the larger cluster. However, it appeared to be more disperse than the antiparallel phylogenetic tree with s = 10. Surprisingly, most of the codons in the clusters did not have template duplication codons, even though they had copies upstream. Their evolution did not agree with the “template duplication mechanism.”
As the degree of similarity (s) S increased, more dispersed clusters appeared in the larger cluster in the parallel phylogenetic tree. As shown in Figure 2(e), the largest cluster is divided into six small clusters with s = 45, where gtc, aac, tct, and tcg remained in isolated branches. A comparison of Figure 2(a), Figure 2(c), and Figure 2(e) shows that the codon aac occupies a basal clade and it did not vary with the degree of similarity (s) in the three phylogenetic trees. The evolutionary relationships among the branches of the three phylogenetic trees were always ordered and stable with different values of s. Most tRNA anticodons in the same clusters had a high match with the “point mutation mechanism” and most had only one base difference from the anticodon sites, which suggests that they evolved by mutating from the same ancestral sequences via point mutations. At the same time, many isoaccepting tRNA codons were distributed in the same cluster and they had a very compact structure. However, not all of the isoaccepting tRNA codons had neighboring relationships, e.g., four codons in the isoaccepting group for Arg were neighboring whereas the other two were far from their branch, which suggested that these two codons may have evolved from neighboring codons. In contrast to the sparse distribution of the parallel phylogenetic tree, the antiparallel phylogenetic tree with s = 45 comprised two large clusters with a more compact branch distribution. One large cluster also appeared to have a laddered rank. Similar results are shown in Figure 2(f) where more isoaccepting tRNA codons had neighbors in the same cluster, e.g., five codons for Leu were adjacent in a large cluster. However, not all of the isoaccepting tRNA codons were allocated to the same cluster. Figure 2(f) shows that evidence for duplication was absent from the entire tree.
As the degree of similarity s increased to 50, the distances of most codon branches in the parallel and antiparallel phylogenetic trees tended toward 1, as shown in Figure 2(g). The parallel phylogenetic trees with s = 50 displayed a laddered rank, but there were few isolated branches and the distances among most branches were similar. There were also many small clusters that contained some codons of isoaccepting tRNAs. Most of these codons showed no evidence point mutations traces at higher levels in the upstream branch. With s > 50, most of the distances for the codons in the parallel phylogenetic trees were equal to 1. There were clear differences in the parallel phylogenetic trees in Figure 2(h), while most of the distances among codon branches in the antiparallel phylogenetic trees with s = 50 were equal to 1, and the distribution of the tree was a laddered rank.
4. Conclusions
We constructed ![]() groups of networks and eight phylogenetic trees of tRNAs based on a point mutation mechanism and a complete duplication mechanism. We analyzed the features of each codon branch and the phylogenetic trees in parallel and antiparallel directions. We found that point mutations had an important role in the evolution of the 62 codon sequence groups. Point mutations were found in the codon sites in the same clusters in four complete parallel phylogenetic trees, whereas there was no evidence for a complementary duplication mechanism in the corresponding antiparallel phylogenetic trees. In addition, many codons of isoaccepting tRNA were found in neighboring clusters or very close clusters. The codons with one site difference were very close together. These results support the hypothesis that new tRNA genes were recruited from one isoacceptor group to another via a point mutation in the sequence.
groups of networks and eight phylogenetic trees of tRNAs based on a point mutation mechanism and a complete duplication mechanism. We analyzed the features of each codon branch and the phylogenetic trees in parallel and antiparallel directions. We found that point mutations had an important role in the evolution of the 62 codon sequence groups. Point mutations were found in the codon sites in the same clusters in four complete parallel phylogenetic trees, whereas there was no evidence for a complementary duplication mechanism in the corresponding antiparallel phylogenetic trees. In addition, many codons of isoaccepting tRNA were found in neighboring clusters or very close clusters. The codons with one site difference were very close together. These results support the hypothesis that new tRNA genes were recruited from one isoacceptor group to another via a point mutation in the sequence.
Surprisingly, most of the codons in the antiparallel phylogenetic trees in the same cluster did not have complementary copies without the point mutation codons, although they were compared in the antiparallel direction. As shown in Figure 3, most codons in the antiparallel phylogenetic tree with s = 45 had no
![]()
Figure 3. Codons in the antiparallel phylogenetic tree where s = 45 and its possible complementary duplication anticodons. The symbol “*” indicates that this anticodon has a copy in the cluster.
complementary codons, although many had point mutation codons in the same cluster. These observations also applied to the other antiparallel trees with different s values. They are marked with a black solid dot (●) in all of the trees and they provide powerful support for the role of point mutations in the evolution of the 64 tRNA codon sequences.
Point mutations occur in the mitochondria tRNA; some positions will cause loss of stability or produce disease. As more and more tRNA sequencing, the mutation of the tRNA gene sequences is studied further, and the evolutionary mechanisms of the RNA will be proven.
Acknowledgements
This paper was supported by the National Science Foundation of Guanxi of China (Grant No.: AE052095) and the National Regional Fund (Grant No.: 11262003) and Guangxi Key Laboratory for the Relativistic Astrophysics.