Facile, Direct Reaction of Benzaldehydes to 3-Arylprop-2-Enoic Acids and 3-Arylprop-2-Ynoic Acids in Aqueous Medium ()
Received 27 March 2016; accepted 31 May 2016; published 3 June 2016

1. Introduction
Cinnamic acids and their derivatives have a wide range of uses. They can have antifungal [1] and/or antimicrobial [2] activity. Applications of cinnamic acid and derivatives can be found in the cosmetic industry, where they have been employed as firming agents in formulations used in skin care products [3] . Additionally, cinnamic acids and derivatives have found use as flavorants [4] , and especially the alkoxylated and hydroxylated cinnamic acids are utilized as stabilizers in the food industry [5] . Also, they are key building blocks in various synthetic preparations in the industry. Cinnamic acid itself is an intermediate in the production of phenylalanine, and thus of aspartame [6] . The authors [7] [8] have communicated their interest in cinnamic acids previously, because some cinnamic acids [7] - [9] and especially dihydroxycinnamic acid derivatives show anti-cancer activity.
Cinnamic acids have been prepared in a number of ways. Among them, the reaction of benzaldehydes with malonic acid is the most common (Knoevenagel reaction with subsequent decarboxylation [10] - [11] ). This strategy has been augmented recently by the reaction of benzaldehydes with Meldrum’s acid [12] . Also, cinnamic acids can be prepared by the reaction of benzaldehydes with acetic anhydride/sodium acetate (Perkin reaction [13] ), and of iodoarenes with acrylates via Heck reaction [14] with subsequent hydrolysis in a two-step sequence. Some of the reactions above give variable yields and others require special catalysts [12] . Therefore, the Wittig olefination reaction of benzaldehydes with stabilized alkoxymethylidenetriphenylphosphoranes remains an alternative in the preparation of cinnamic acids, and so has been used extensively, too [15] .
Conventional Wittig reactions with stabilized phosphoranes have been carried under elevated temperature in organic solvents such as THF and benzene, sometimes under acid catalysis (Bestmann variation) [16] . In more recent times, so-called non-classical reaction conditions [17] have been forwarded for the Wittig olefination, such as carrying out reactions under solventless [18] conditions or in ionic liquids [19] . Often, this has been combined with microwave irradiation or ultrasonication. Also, water soluble phosphoranes [20] - [22] have been developed to carry out the reactions in aqueous solutions. Afterwards, it was seen that olefination reactions could also be performed in water with stabilized and semi-stabilized, non-water soluble phosphoranes [23] - [34] , with one communication on a Wittig reaction utilizing semi-stabilized and non-stabilized phosphoranes in a biphasic mixture under phase transfer conditions (PTC) [35] . Nevertheless, almost all of these cases involve extractive work-up and often chromatographic separations using organic solvents. Mostly, such work-up utilizes greater amounts of organic solvent than does the reaction itself, if it were run in organic medium. Before, the authors had developed a biphasic reaction system of water and hexane for the Wittig olefination with stabilized and semi-stabilized phosphoranes [36] . Here, the reaction proceeds mostly at the solvent interface [37] , giving the product after simple evaporation of the hexane phase, where the organic solvent can be recycled. Nevertheless, possibilities of using such a strategy are rare.
In the following, a methodology is introduced which involves a one-pot olefination-hydrolysis sequence, where the unsaturated carboxylic acid is the final product, which can be isolated by acidification of the reaction medium, and, in the case of the cinnamic acid products, by simple filtration. The reaction sequence is well- suited for educational laboratories also, and has been shown to give reproducible good yields with our undergraduate students.
2. Experimental
2.1. Chemicals and Instruments
Experimental
General Remarks. Melting points were measured on a Stuart SMP 10 melting point apparatus and are uncorrected. Infrared spectra were measured with a Thermo/Nicolet Nexus 470 FT-IR ESP spectrometer. 1H and 13C NMR spectra were recorded with a Varian 400 NMR spectrometer (1H at 395.7 MHz, 13C at 100.5 MHz). The assignments of the carbon signals were aided by DEPT 90 and DEPT 135 experiments (DEPT = Distortionless Enhancement by Polarisation Transfer). The chemical shifts are relative to TMS (solvent CDCl3, unless otherwise noted). Mass spectra were measured with a JMS-01-SG-2 spectrometer, and with an Agilent QTOF 6540 UHD. Column chromatography, where necessary, was performed on silica gel (S, 0.063 mm - 0.1 mm, Riedel de Haen and Merck grade 9385).
2-Methoxybenzaldehyde (1a), 2,4-dimethoxybenzaldehyde (1g) and 4-benzyloxybenzaldehyde (1x) were prepared from the corresponding, commercially available hydroxybenzaldehydes (KOH, DMSO, CH3I or PhCH2Br) [38] . Benzaldehydes 1n and 1o were synthesized by bromination (NBS, DMF, rt) of 2-hydroxy- and 4-hydroxybenzaldehydes, respectively, followed by alkylation (KOH, DMSO, CH3I). The other benzaldehydes were acquired commercially. The 4-haloacetophenones 7b-d and 1-acetylanthracene (8) were obtained by acetylation (AcCl, AlCl3) of the parent acetophenones and anthracene, respectively. Phosphoranes 2a [39] and 2b [40] were prepared according to literature procedures.
20 and 22 were prepared by Wittig olefination, starting from 2-benzyloxybenzaldehyde and benzoylmethylidenetriphenylphosphorane and from 2-benzyloxycinnamaldehyde (24) and toluoylmethylidenetriphenylphosphorane.
2.2. General Procedure A
(E)-Cinnamic acid (3f). To a mixture of benzaldehyde (1f, 1.60 g, 15.1 mmol) and ethoxycarbonylmethylidenetriphenylphosphorane (2a, 6.82 g, 19.5 mmmol) was given an aq. NaOH solution (9.1 w%, 3.5 g (87.5 mmol) NaOH in 35 mL H2O) and the resulting suspension was stirred at 75˚C for 23 h. Triphenylphosphine oxide was filtered off the cooled solution. Thereafter, the filtrate was acidified carefully with 15 w% aq. HCl. The resulting suspension was cooled and filtered. The solid obtained was dried in air to give cinnamic acid (3f, 1.89 g, 77%) as a colorless solid; mp. 132˚C [Lit. 132˚C - 135˚C [41] ]; υmax (KBr/cm−1) 1690 (C=O), 1631, 1454, 1312, 1290, 1225, 771, 714; δH (270 MHz, DMSO-d6) 6.51 (1H, d, 3J = 16.0 Hz), 7.39 (2H, m), 7.57 (1H, d, 3J = 16.0 Hz), 7.59 - 7.66 (3H, m), 12.35 (1H, bs); δC (67.8 MHz, DMSO-d6) 119.7, 128.6 (2C), 129.3 (2C), 130.7, 134.7, 144.4, 168.0; MS (EI, 70 eV) m/z (%) 148 (M+) (100), 77 (40).
(E)-2-Methoxycinnamic acid (3a) (Procedure A). Colorless solid; mp. 187˚C [Lit. 183˚C - 186˚C [41] ]; νmax (KBr/cm−1) 1682 (C=O), 1620 (C=C), 1489, 1463, 1426, 1332, 1282, 1248, 1220 (C-O), 1025, 994, 941, 755, 696, 596, 567; δH (400 MHz, DMSO-d6) 3.84 (3H, s, OCH3), 6.48 (1H, d, 3J = 16.0 Hz), 6.96 (1H, dd, 3J = 7.6 Hz, 3J = 7.2 Hz), 7.06 (1H, d, 3J = 8.4 Hz), 7.36 - 7.40 (1H, m), 7.64 - 7.66 (1H, m), 7.81 (1H, d, 3J = 16.0 Hz); δC (100.5 MHz, DMSO-d6) 56.1, 112.2, 119.7, 121.2, 122.9, 128.9, 132.2, 139.1, 158.2, 168.3; MS (EI, 70 eV) m/z (%) = 178 (M+, 89), 147 (M+-OCH3, 100).
(E)-4-Methoxycinnamic acid (3b) (Procedure A). Colorless solid; mp. 173˚C [Lit. 173˚C [41] ]; νmax (KBr/ cm−1) 1694 (C=O), 1596 (C=C), 1336, 826, 685, 566, 527, 513; δH (400 MHz, CDCl3) 3.85 (3H, s, OCH3), 6.33 (1H, d, 3J = 16.0 Hz), 6.92 (2H, d, 3J = 8.8 Hz), 7.51 (2H, d, 3J = 8.8 Hz), 7.75 (1H, d, 3J = 16.0 Hz); δC (100.5 MHz, CDCl3) 55.4 (OCH3), 114.4 (2C, CH), 114.6 (CH), 126.8 (Cquat), 130.1 (2C, CH), 146.7 (CH), 161.7 (Cquat), 172.2 (Cquat, CO); MS (70 eV, EI) m/z (%) 178 (M+, 65).
(E)-4-Ethoxycinnamic acid (3c) (Procedure A). Colorless solid, mp. 196˚C [Lit. mp. 195˚C - 199˚C [41] ]; δH (400 MHz, DMSO-d6) 1.30 (3H, t, 3J = 7.2 Hz, CH3), 4.02 (2H, q, 3J = 7.2 Hz, OCH2), 6.34 (1H, d, 3J = 16.0 Hz), 6.92 (2H,. d, 3J = 8.4 Hz), 7.51 (1H, d, 3J = 16.0 Hz), 7.59 (2H, d, 3J = 8.4 Hz); δC (100.5 MHz, DMSO-d6) 15.0 (CH3), 63.7 (CH2), 115.2 (2C, CH), 116.8 (CH), 127.1 (Cquat), 130.4 (2C, CH), 144.2 (CH), 160.7 (Cquat), 168.3 (Cquat, CO); MS (EI, 70 eV) m/z (%) = 192 (M+, 61), 146 (54), 118 (100).
(E)-4'-Propoxycinnamic acid (3d) (Procedure A). Colorless solid, mp. 155˚C [Lit. 155˚C [42] ]; δH (400 MHz, DMSO-d6) 0.96 (3H, t, 3J = 7.2 Hz, CH3), 1.70 (qt, 3J = 7.2 Hz, 3J = 6.4 Hz), 3.95 (2H, t, 3J = 6.4 Hz), 6.34 (1H, d, 3J = 16.0 Hz), 6.94 (2H, d, 3J = 8.8 Hz), 7.51 (1H, d, 3J = 16.0 Hz), 7.59 (2H, d, 3J = 8.8 Hz); δC (100.5 MHz, DMSO-d6) 14.1, 19.1, 67.8, 115.2 (2C), 116.8, 127.1, 130.4, 144.2, 160.9, 168.3.
(E)-4'-(N,N-Dimethylamino)cinnamic acid (3e) (Procedure A). Colorless solid, mp. 226˚C; [Lit. 227 - 228˚C [41] ]; νmax (KBr/cm−1) 2922 (CH), 1679 (C=O), 1598 (C=C), 1230, 1202, 815; δH (400 MHz, DMSO-d6) 2.94 (6H, s, 2 CH3), 6.19 (1H, d, 3J = 16.0 Hz), 6.68 (2H, d, 3J = 9.2 Hz), 7.43 (1H, d, 3J = 16.0 Hz), 7.45 (2H, d, 3J = 9.2 Hz), 11.93 (bs, 1H, OH); δC (100.5 MHz, CDCl3) 39.4 (2C, CH3), 111.8 (2C, CH), 113.1 (CH), 121.6 (Cquat), 129.6 (2C, CH), 144.6 (CH), 151.6 (Cquat), 168.0 (Cquat, CO); MS (EI, 70 eV) m/z (%) 191 (M+, 100), 147 (17).
(E)-2,4-Dimethoxycinnamic acid (3g) (Procedure A). Colorless solid, mp. 191˚C; [Lit. 192˚C - 194˚C [41] ]; δH (400 MHz, DMSO-d6/CDCl3) 3.81 (3H, s, OCH3), 3.86 (3H, s, OCH3), 6.33 (1H, d, 3J = 16.0 Hz), 6.51 (2H, m), 7.47 (1H, d, 3J = 8.0 Hz), 7.80 (1H, d, 3J = 16.0 Hz); δH (100.5 MHz, DMSO-d6/CDCl3) 55.2 (OCH3), 55.3 (OCH3), 98.1 (CH), 105.9 (CH), 115.9 (Cquat), 116.3 (CH), 129.6 (CH), 138.8 (CH), 159.4 (Cquat), 162.7 (Cquat), 168.3 (Cquat, CO).
(E)-2,5-Dimethoxycinnamic acid (3h) (Procedure A). Colorless solid; mp. 149 oC; [Lit. 148˚C - 150˚C [41] ]; νmax (KBr/cm−1) 1697 (C=O), 1627 (C=C), 1581, 1503, 1430, 1323, 1228 (C-O), 1189 (C-O), 1052, 1019, 988, 941, 858, 788, 700, 629, 537, 487, 456; δH (400 MHz, DMSO-d6) 3.72 (3H, s, OCH3), 3.78 (3H, s, OCH3), 6.53 (1H, d, 3J = 16.4 Hz), 6.97 (1H, d, 3J = 6.4 Hz), 6.99 (1H, d, 3J = 6.4 Hz), 7.21 (1H, d, 4J = 3.2 Hz), 7.78 (1H, d, 3J = 16.4 Hz); δC (100.5 MHz, CDCl3) 56.0 (OCH3), 57.0 (OCH3), 112.5, 113.5, 117.8, 118.0, 123.6, 142.2, 153.2 (Cquat), 153.6 (Cquat), 173.0 (Cquat, CO); MS (EI, 70 eV) m/z (%) = 208 (M+, 100), 177 (M+-OCH3, 33).
4-Bromo-2,5-dimethoxycinnamic acid (3i) (Procedure A) as a mixture of E- and Z-isomers (72/28), pale yellow solid, mp. 177˚C - 180˚C (for the E-/Z-mixture); νmax (KBr/cm−1) [E/Z-mixture] 3502 - 2829 (bs, OH), 1687 (C=O), 1677 (C=O), 1629 (C=C), 1565, 1499, 1441, 1392, 1329, 1255, 1207, 1052, 987, 917, 867, 822, 739, 696, 638, 617, 541; (Z)-isomer δH (400 MHz, CDCl3) 3.80 (3H, s, OCH3), 3.84 (3H, s, OCH3), 6.01 (1H, d, 3J = 12.8 Hz), 7.07 (1H, s), 7.23 (1H, d, 3J = 12.8 Hz), 7.39 (1H, s); δC (100.5 MHz, CDCl3) 56.2 (OCH3), 56.3 (OCH3), 113.5 (Cquat), 114.8 (CH), 115.8 (CH), 119.0 (CH), 123.0 (Cquat), 140.4 (CH), 149.3 (Cquat), 151.8 (Cquat), 170.9 (Cquat, CO); (E)-isomer δH (400 MHz, CDCl3) 3.85 (3H, s, OCH3), 3.89 (3H, s, OCH3), 6.51 (1H, d, 3J = 16.0 Hz), 7.03 (1H, s), 7.13 (1H, s), 8.00 (1H, d, 3J = 16.0 Hz); δC (100.5 MHz, CDCl3) 56.3 (OCH3), 56.8 (OCH3), 111.6 (CH), 115.3 (Cquat), 116.9 (CH), 118.0 (CH), 122.6 (Cquat), 141.5 (CH), 150.2 (Cquat), 153.0 (Cquat), 172.2 (Cquat, CO); MS (EI, 70 eV) m/z (%) = 288 ([81BrM+, 11), 286 ([79BrM+, 13).
(E)-4-Methylcinnamic acid (3j) (Procedure A). Colorless solid, mp. 195˚C - 199˚C [Lit. 196˚C - 198˚C [41] ]; νmax (KBr/cm−1) 3550 - 2330 (bs, OH), 1678 (C=O), 1624 (C=C), 1423, 1312, 1285, 1223, 987, 944, 812, 687, 554, 495; δH (400 MHz, CDCl3) 2.38 (3H, s, CH3), 6.40 (1H, d, 3J = 16.0 Hz), 7.21 (2H, d, 3J = 8.0 Hz), 7.45 (2H, d, 3J = 8.0 Hz), 7.76 (1H, d, 3J = 16.0 Hz); δH (400 MHz, DMSO-d6) 2.33 (3H, s, CH3), 6.49 (1H, d, 3J = 16.0 Hz), 7.23 (2H, d, 3J = 8.4 Hz), 7.57 (2H, d, 3J = 8.4 Hz), 7.58 (1H, d, 3J = 16.0 Hz), 12.3 (1H, bs, OH); δC (100.5 MHz, DMSO-d6) 30.0 (CH3), 118.1 (CH), 128.2 (2C, CH), 129.6 (2C, CH), 131.6 (Cquat), 140.2 (Cquat), 144.0 (CH), 167.6 (Cquat, CO); MS (EI, 70 eV) m/z (%) = 162 (100) (M+), 115 (43).
(E)-3-Chlorocinnamic acid (3k) (Procedure A). Colorless crystals, mp. 165˚C; [Lit. 161˚C - 164˚C [41] ]; νmax (KBr/cm−1) 1677 (C=O), 1632 (C=C), 1430, 1323, 1301, 1283, 1230, 1200, 943, 864, 727, 680, 669, 597, 550; δH (400 MHz, DMSO-d6) 6.59 (1H, d, 3J = 16.4 Hz), 7.41 - 7.45 (2H, m), 7.55 (1H, d, 3J = 16.4 Hz), 7.63 - 7.65 (1H, m), 7.78 - 7.79 (1H, m); 13C NMR (100.5 MHz, DMSO-d6) 121.4, 127.2, 128.3, 130.2, 131.1, 134.2, 137.0, 142.7, 167.8; MS (EI, 70 eV) m/z (%) = 182 ([81Cl]M+, 100), 181 ([79ClM+], 77), 147 (36).
3-Bromocinnamic acid (3L) (Procedure A). Colorless solid; mp. 170˚C [Lit. 177˚C [41] ]; νmax (KBr/cm−1) 3050 - 2550 (bs, OH), 2972 (C-H), 1694 (C=O), 1629 (C=C), 1424, 1318, 1219 (C-O), 977, 936, 784, 720, 664, 542; δH (400 MHz, DMSO-d6) 6.57 (1H, d, 3J = 16.4 Hz), 7.35 (1H, dd, 3J = 8.0 Hz, 3J = 8.0 Hz), 7.52 - 7.62 (2H, m), 7.52 (1H, d, 3J = 16.4 Hz), 7.67 (1H, bd, 3J = 8.0 Hz), 7.89 (1H, m); δC (100.5 MHz, DMSO-d6) 121.3, 122.7, 127.5, 131.1, 131.4, 133.2, 137.2, 142.7, 167.8 (CO); MS (EI, 70 eV) m/z (%) = 228 (95) ([81Br]M+), 226 (100) ([79Br]M+), 147 (61).
(E)-3-Fluorocinnamic acid (3m) (Procedure A).Colorless solid; mp. 168˚C - 169˚C [Lit. 167˚C - 170˚C [41] ]; νmax (KBr/cm−1) 1693 (C=O), 1632, 1588, 1295, 1249, 940, 859, 782, 552; δH (400 MHz, DMSO-d6) 6.57 (1H, d, 3J = 16.0 Hz), 7.19 - 7.24 (1H, m), 7.40 - 7.56 (3H, m), 7.50 (1H, d, 3J = 16.0 Hz); δC (100.5 MHz, DMSO-d6) 114.8 (2JCF = 21.7 Hz), 117.3 (2JCF = 21.0 Hz), 121.3, 125.1, 131.3 (3JCF = 8.2 Hz), 137.2 (3JCF = 8.2 Hz), 143.0, 162.8 (1JCF = 243 Hz), 167.9 (CO); MS (EI, 70 eV) m/z (%) = 166 (100) (M+), 149 (30), 121 (33).
(E)-3,5-Dibromo-4-methoxycinnamic acid (3n) [43] (Procedure A). Colorless solid; mp. 198˚C. νmax (KBr/ cm−1) 3100 - 2597 (bs, OH), 1703 (C=O), 1633 (C=C), 1472, 1424, 1268, 1217, 978, 851, 747; δH (400 MHz, CDCl3) 3.92 (3H, s, OCH3), 6.37 (1H, d, 3J = 16.4 Hz), 7.59 (1H, d, 3J = 16.4 Hz), 7.69 (2H, s); δC (100.5 MHz, CDCl3) 60.8 (OCH3), 118.8 (2C, Cquat), 119.0 (CH), 132.3 (2C, CH), 143.3 (CH), 155.8 (Cquat), 170.9 (Cquat, CO).
(E)-2-Bromo-4,5-dimethoxycinnamic acid (3o) (Procedure A).Colorless solid, mp. 240˚C - 244˚C [Lit. 246˚C - 247˚C [44] ]; νmax (KBr/cm−1) 3496 - 2578 (bs, OH), 1697 (C=O), 1596 (C=C), 1512, 1390, 1310, 1267, 1206, 1163, 1027, 969, 854, 814; δH (400 MHz, CDCl3) 3.87 (3H, s, OCH3), 3.90 (3H, s, OCH3), 6.55 (1H, d, 3J = 16.0 Hz), 6.58 (1H, s), 6.77 (1H, s), 7.60 (1H, d, 3J = 16.0 Hz); δC (100.5 MHz, CDCl3) 56.0 (OCH3), 56.2 (OCH3), 114.1 (CH), 115.4 (Cquat), 118.3 (CH), 122.0 (CH), 131.6 (Cquat), 144.6 (CH), 148.5 (Cquat), 150.3 (Cquat), 167.7 (Cquat, CO). Found: C, 45.70%; H, 3.86%. Calcd. for 46.02%; H, 3.86%.
(2E,4E)-5-Phenylpenta-2,4-dienoic acid (3p) (Procedure A). Pale yellow solid, mp. 165˚C - 166˚C [Lit. mp. 165˚C [45] ]; νmax (KBr/cm−1) 3500 - 2850 (bs, OH), 1678 (C=O), 1609 (C=C), 1582, 1285; δH (400 MHz, CDCl3) 6.00 (1H, d, 3J = 15.0 Hz), 6.90 - 6.96 (2H, m), 7.21 - 7.56 (6H, m); δC (100.5 MHz, CDCl3) 120.1, 126.1, 127.3, 128.9, 129.3, 135.9, 141.7, 146.9, 171.8; MS (EI, 70 eV) m/z (%) = 174 (21) (M+), 129 (100).
(E)-3-(Thien-2-yl)acrylic acid (3q) (Procedure A). Colorless solid, mp. 145˚C; [Lit. 145˚C - 148˚C [41] ]. νmax (KBr/cm−1) 3200 - 2500 (bs, OH), 1677 (C=O), 1615 (C=C), 1410, 1308, 1274, 1244, 1218, 1190, 972, 925, 860, 834, 715, 598, 531; δH (400 MHz, CDCl3) 6.24 (1H, d, 3J = 15.6 Hz), 7.08 (1H, dd, 3J = 5.2 Hz, 3J = 4.0 Hz), 7.31 (1H, d, 3J = 4.0 Hz),7.43 (1H, d, 3J = 5.2 Hz), 7.88 (1H, d, 3J = 15.6 Hz); δC (100.5 MHz, CDCl3) 115.9 (CH), 128.2 (CH), 129.3 (CH), 131.7 (CH), 139.2 (Cquat), 139.4 (CH), 172.2 (Cquat, CO); MS (FAB, 3-nitrobenzyl alcohol) m/z (%) = 155 (17) (MH+).
(E)-5-Bromothien-2-ylacrylic acid (3r) (Procedure A). Colorless solid, mp. 207˚C [Lit. 209˚C - 210˚C [46] ]; νmax (KBr/cm−1) 3430 - 2597 (bs, OH), 1688 (C=O), 1615 (C=C), 1433, 1407, 1298, 1268, 1211, 966, 795, 533; δH (400 MHz, DMSO-d6) 6.11 (1H, d, 3J = 16.0 Hz), 7.22 (1H, d, 3J = 4.4 Hz), 7.29 (1H, d, 3J = 4.4 Hz), 7.62 (1H, d, 3J = 16.0 Hz); δC (100.5 MHz, DMSO-d6) 115.4, 118.6, 132.3, 132.9, 136.3, 141.1, 167.6.
(E)-3,4-Dimethoxycinnamic acid (3s) (Procedure A). Colorless solid, mp. 183˚C [Lit. 181˚C - 183˚C [41] ]; νmax (KBr/cm−1) 3300 - 2500 (bs, OH), 1677 (C=O), 1624, 1596, 1517, 1463, 1426, 1340, 1250, 1140, 1025, 975, 840, 579, 536; δH (400 MHz, DMSO-d6) 3.78 (3H, s, OCH3), 3.80 (3H, s, OCH3), 6.41 (1H, d, 3J = 16.0 Hz), 6.86 (1H, d, 3J = 8.0 Hz), 7.10 (1H, s), 7.13 (1H, d, 3J = 8.0 Hz), 7.50 (1H, d, 3J = 16.0 Hz), 12.19 (1H, bs, OH); δC (100.5 MHz, DMSO-d6) 56.0, 56.1, 110.7, 112.0, 117.2, 123.1, 127.5, 144.6, 149.4, 151.2, 168.3; MS (EI, 70 eV) m/z (%) = 208 (M+, 100), 193 (M+-CH3, 21).
(E)-3,4-Diethoxycinnamic acid (3t) (Procedure A). Colorless solid, mp. 154˚C [Lit. 156˚C [47] ] δH (400 MHz, CDCl3) 1.46 (6H, t, 3J = 6.8 Hz, 2 CH3), 4.12 (2H, q, 3J = 6.8 Hz, OCH2), 4.13 (2H, q, 3J = 6.8 Hz, OCH2), 6.28 (1H, d, 3J = 16.0 Hz), 6.85 (1H, d, 3J = 8.0 Hz), 7.07 (1H, s), 7.08 (1H, d, 3J = 8.0 Hz), 7.70 (1H, d, 3J = 16.0 Hz); δC (100.5 MHz, CDCl3) 14.6, 14.7, 64.5, 64.6, 111.9, 112.6, 114.6, 123.1, 126.9, 147.1, 148.8, 151.3, 172.4.
(E)-3,4-(Methylenedioxy)-cinnamic acid (3u) (Procedure A). Mp. 214˚C [Lit. 242˚C - 244˚C [41] ]; ν (KBr/cm−1) 3200 - 2500 (bs, OH), 1693 (C=O), 1624 (C=C), 1495, 1446, 1314, 1251, 1100, 1036, 924, 805; δH (400 MHz, DMSO-d6) 6.08 (2H, s), 6.39 (1H, d, 3J = 15.6 Hz), 6.95 (1H, d, 3J = 8.0 Hz), 7.16 (1H, dd, 3J 8.0 Hz, 4J = 1.6 Hz), 7.37 (1H, d, 4J = 1.6 Hz), 7.51 (1H, d, 3J = 15.6 Hz); δC (100.5 MHz, DMSO-d6) 101.4, 106.6, 108.4, 117.0, 124.5, 128.6, 143.7, 147.9, 149.0, 167.7; MS (EI, 70 eV) m/z (%) = 192 (M+, 100), 145 (13).
(E)-2-Chlorocinnamic acid (3v) (Procedure A). Colorless solid; mp. 200˚C [Lit. mp. 208˚C - 210˚C [41] ]; νmax (KBr/cm−1) 3050 - 2500 (bs, OH), 2971 (C-H), 1687 (C=O), 1622 (C=C), 1427, 1281, 1224, 1041, 978, 921, 727, 595, 560; for E-3v: δH (400 MHz, DMSO-d6) 6.57 (1H, d, 3J = 16.0 Hz), 7.51 (1H, dd, 3J = 7.6 Hz, 4J = 1.6 Hz), 7.35 ? 7.45 (2H, m), 7.84 (1H, d, 3J = 16.0 Hz), 7.89 (1H, dd, 3J = 7.6 Hz, 4J = 2.0 Hz); δC (100.5 MHz, DMSO-d6) 122.7, 128.3, 128.6, 130.4, 132.2 (2C), 134.0, 139.2, 167.7 (Cquat, CO); for Z-3v: δH (400 MHz, DMSO-d6) 6.10 (1H, d, 3J = 12.0 Hz), 7.03 (1H, d, 3J = 12.0 Hz), 7.26 - 7.45 (4H, m); δC (100.5 MHz, DMSO- d6) 124.2, 127.1, 129.3, 131.1, 132.3 (2C), 134.5, 138.5, 167.2 (Cquat, CO); MS (EI, 70 eV) m/z (%) = 182 (15) ([35Cl]M+), 147 (100).
Ethyl (E)-4-nitrocinnamate (3w) and (E)-4-nitrocinnamic acid (3x). A mixture of 4-nitrobenzaldehyde (1w, 1.51 g, 10 mmol) and phosphorane 2a (4.87 g, 14 mmol) in chloroform (0.5 mL) was heated to 120˚C for 45 min. The cooled solution is subjected directly to column chromatography on silica gel (CHCl3/ether/hexane 1:1:1) to give 3w (1.97 g, 89%) as a light yellow solid; mp. 140˚C; [Lit. 138˚C - 140˚C [41] ]; νmax (KBr/cm−1) 3106, 3079 (C-Harom.), 2983 (C-Haliph), 2939 (C-Haliph), 1712 (C=O), 1645, 1594, 1340 (NO2(s)), 1189, 1177, 1110, 1029, 1189, 1171, 1110, 1028, 978, 845, 759; δH (400 MHz, CDCl3) 1.35 (3H, t, 3J = 7.2 Hz), 4.29 (2H, q, 3J = 7.2 Hz), 6.55 (1H, d, 3J = 16.4 Hz), 7.70 (1H, d, 3J = 16.4 Hz), 7.66 (2H, d, 3J = 8.8 Hz), 8.25 (2H, d, 3J = 8.8 Hz). A solution of 3w (1.95 g, 8.82 mmol) in a mixture of aq. NaOH (10w%, 50 mL) and methanol (10 mL) was heated at 65˚C for 12h. Thereafter, the solution was filtered, and the filtrate was acidified with half-conc. aq. HCl. The formed precipitate was filtered, washed with water (3 × 20 mL) and air-dried to give 3x (1.62 g, 95%) as a light yellowish solid; mp. 287˚C [Lit. mp. 289˚C [40] ]; νmax (KBr/cm−1) 3500 - 2499 (bs, OH), 1686 (C=O), 1628 (C=C), 1522 (NO2(as)), 1426, 1350 (NO2(s)), 1308, 1283, 1222, 985, 836, 716; δH (400 MHz, DMSO-d6) 6.69 (1H, d, 3J = 16.0 Hz), 7.64 (1H, d, 3J = 16.0 Hz), 7.92 (2H, d, 3J = 8.8 Hz), 8.20 (2H, d, 3J = 8.8 Hz); δC (100.5 MHz, DMSO-d6) 124.4 (2C, CH), 129.7 (2C, CH), 123.9 (CH), 141.1 (Cquat), 141.8 (CH), 148.4 (Cquat), 167.5 (Cquat, CO); MS (EI, 70 eV) m/z (%) 193 (M+, 65), 176 (18), 147 (15).
(Z)-4-Benzyloxycinnamic acid (3y) (Procedure A). Colorless solid; mp. 168˚C; δH (400 MHz, DMSO-d6) 5.11 (2H, s, OCH2), 5.79 (1H, d, 3J = 12.8 Hz), 6.80 (1H, d, 3J = 12.8 Hz), 6.98 (2H, d, 3J = 8.8 Hz), 7.40 (2H, d, 3J = 8.4 Hz), 7.57 (2H, d, 3J = 8.4 Hz), 7.67 (2H, d, 3J = 8.8 Hz); δC (100.5 MHz, DMSO-d6) 68.8 (OCH2), 114.7 (2C, CH), 118.9, 121.4, 128.0, 129.2, 130.3 (2C, CH), 131.8 (2C, CH), 132.4 (2C, CH), 136.8, 141.2, 159.2 (Cquat), 168.0 (Cquat, CO); MS (Ion trap): m/z 255 (MH+).
Ethyl 3(E)-(9-anthranyl)propenoate (3z-Et). A mixture of 9-anthranylcarbaldehyde (1y, 1.0 g, 4.85 mmol) and ethoxycarbonylmethylidenephosphorane (2a, 2.36 g, 6.79 mmol) is heated at 130˚C for 3h. Thereafter, an additional amount of 2a (348 mg, 1.0 mmol) is added and the reaction mixture heated for another hour at 135˚C. The cooled solution is subjected directly to column chromatography on silica gel (eluent: MtBE/CHCl3/hexane 1:1:7) to give 3z-Et as a yellow-orange solid (1.23 g, 92%), mp. 80˚C [Lit. 80˚C [48] ]; δH (400 MHz, CDCl3) 1.35 (3H, t, 3J = 7.2 Hz), 4.31 (2H, q, 3J = 7.2 Hz, OCH2), 6.36 (1H, d, 3J = 16.0 Hz), 7.43 (4H, m), 7.95 (2H, m), 8.17 (2H, m), 8.39 (1H, s), 8.57 (1H, d, 3J = 16.0 Hz); δC (100.5 MHz, CDCl3) 14.5, 61.0, 125.2, 125.4, 127.2, 128.2, 128.8, 129.3, 129.4, 131.2, 141.9, 166.5; MS: Found: 299.1040 (C19H16O2+Na)+; Calcd. for C19H16O2Na: 299.1048.
Alternatively, to the reaction mixture of 1y and 2a, aq. NaOH (10 w%, 15 mL) was added after 4h, and the resulting solution was kept at reflux for 14 h. Thereafter, the solution was cooled and the precipitate was filtered off. The precipitate which contains both triphenylphosphine oxide and sodium 3(E)-(9-anthranyl)propenoate (3z-Na, see below) was washed diligently with hot water, to dissolve the remainder of the salt. Acidification of the cool filtrate with half conc. aq. HCl provides a precipitate, which was filtered, washed with water and dried to yield 3-(9-anthranyl)propenoic acid (3z) (1.02 g, 85%) as a yellow solid; mp. 242˚C [Lit. mp. 245˚C [49] ]; νmax (KBr/cm−1) 3500 - 2495 (bs, OH), 3424, 3051 (C-Harom.), 3003 (C-Harom.), 2898 (C-Haliph), 1690 (C=O), 1630, 1425, 1307, 1200, 725; δH (400 MHz, DMSO-d6) 6.32 (1H, d, 3J = 16.4 Hz), 7.57 (4H, m), 8.12 (4H, m), 8.46 (1H, d, 3J = 16.4 Hz), 8.64 (1H, s), 12.75 (bs, OH); δC (100.5 MHz, DMSO-d6) 125.3 (CH), 126.0 (CH), 127.2 (CH), 128.4 (Cquat), 128.8 (Cquat), 129.1 (CH), 129.3 (CH), 129.5 (Cquat), 131.2 (CH), 141.0 (CH), 157.3 (Cquat, CO).
When heated with 10 w% aq. NaOH, 3-(9-anthranyl)propenoic acid (3z) gives sodium 3(E)-(9-anthranyl) propenoate (3z-Na) as golden, shiny leaflets νmax (KBr/cm−1) 3620 - 2850 (bs, OH), 3611 (v), 1635, 1540 (s), 1441, 1392, 1285, 991, 881, 734; δH (400 MHz, CDCl3) 6.14 (1H, d, 3J = 16.0 Hz), 7.50 (4H, m), 7.90 (1H, d, 3J = 16.0 Hz), 8.07 (2H, m), 8.20 (2H, m), 8.50 (1H, s).
4-Nitrobenzoic acid (4) and 4-nitrobenzyl alcohol (5). 4-Nitrobenzaldehyde (1w, 1.51 g, 10 mmol) and phosphorane 2a (4.87 g, 14 mmol) was given to an aq. NaOH solution (9.1 w%, 3.5 g (87.5 mmol) NaOH in 35 mL H2O) and the resulting suspension was stirred at 75˚C for 20 min. With the addition of 4-nitrobenzaldehyde, the solution turned dark-red. The solution was filtered, and the filtrate was extracted with dichloromethane (3 × 25 mL). Then, the aqueous solution was acidified with half-conc. aq. HCl to give 4-nitrobenzoic acid (4) (800 mg, 48%) as a colorless solid, mp. 237˚C [Lit. 237˚C - 240˚C [41] ]; ν (KBr/cm−1) 3065 (OH), 2672, 1695 (C=O), 1609 (C=C), 1543, 1433; δH (400 MHz, CDCl3) 8.13 (2H, d, 3J = 9.2 Hz), 8.28 (2H, d, 3J = 9.2 Hz); δC (100.5 MHz, DMSO-d6) 123.5 (2C, CH), 131.1 (2C, CH), 136.4 (Cquat), 150.5 (Cquat), 166.8 (Cquat, CO). Thereafter the filter cake of the first filtration was taken up in CHCl3 (50 mL) and combined with the CH2Cl2 extract. The combined extract was dried over anhydrous MgSO4 and concentrated in vacuo. The residue was subjected to column chromatography on silica gel (CHCl3-hexane-ether 1:1:1) to give 4-nitrobenzyl alcohol (5, 540 mg, 35%) as a colorless solid, mp. 93˚C [Lit. 92˚C - 94˚C [48] ]; ν (KBr/cm−1) 3550 (bs, OH), 2954 (C-H), 2925 (C-H), 1508 (NO2(a)), 1458 (NO2(s)); δH (400 MHz, CDCl3) 2.11 (1H, t, 3J = 5.6 Hz, OH), 4.83 (2H, d, 3J = 5.6 Hz), 7.52 (2H, cd, 3J = 8.8 Hz), 8.21 (2H, cd, 3J = 8.8 Hz); δH (400 MHz, CDCl3) 64.0 (CH2), 123.7 (2C, CH), 127.1 (2C, CH), 147.3 (Cquat), 148.4 (Cquat); MS (EI, 70 eV) m/z (%) 153 (55, M+), 77 (100).
(E)-4-Hydroxycinnamic acid (6) [50] . A mixture of 1z (835 mg, 6.84 mmol) and 2a (3.35 g, 9.63 mmol) was heated at 135˚C for 1h. Thereafter, aq. NaOH (10 w%, 10 mL) was added to the cooled mixture, and it was stirred for 12 h at rt. The precipitated triphenylphosphine oxide was filtered off, and the filter-cake was washed with water (2 × 15 mL). The filtrate, combined with the washings, was acidified with half-conc. aq. HCl. The formed precipitate was filtered, washed with water ( 2 × 15 mL) and air-dried to yield 6 (850 mg, 74%) as a colorless solid, mp. 211˚C - 212˚C; IR (KBr/cm−1) νmax 3379 (bs, OH), 1672 (C=O), 1628, 1602 (C=C), 1512, 1449, 1313, 1244 (C-O), 1214 (C-O), 1172, 978, 832, 690; δH (400 MHz, DMSO-d6) 6.23 (1H, d, 3J = 16.0 Hz), 6.76 (2H, d, 3J = 8.8 Hz), 7.19 (1H, d, 3J = 16.0 Hz), 7.34 (2H, d, 3J = 8.8 Hz); δH (100.5 MHz, DMSO-d6) 115.0 (CH), 115.9 (2C, CH), 127.2(Cquat), 129.3 (2C, CH), 138.9 (CH), 158.7 (Cquat), 171.0 (Cquat, CO).
While general procedure A was used for the preparation of 8b, 8d/8e, and 8f, the E- and Z-isomers of 8b and 8f and compound 8d and 8e were separated by column chromatography on silica gel (CHCl3/ether 10:1) and crystallized from a mixture of CHCl3-hexane (1:4).
(E)-3-Phenylbut-2-enoic acid (8a) (Procedure A). Colorless solid, mp. 129˚C - 131˚C [Lit. 131˚C [51] ]; δH (400 MHz, DMSO-d6) 2.49 (3H, d, 4J = 1.6 Hz), 6.10 (1H, q, 4J = 1.6 Hz), 7.40 - 7.42 (3H, m), 7.55 - 7.57 (2H, m), 12.27 (1H, bs, OH); δC (100.5 MHz, DMSO-d6) 17.7 (CH3), 117.9 (CH), 126.6 (2C, CH), 129.1 (2C, CH), 129.5 (CH), 141.9 (Cquat), 154.2 (Cquat), 168.0 (Cquat, CO).
(Z)-3-(4-Chlorophenyl)but-2-enoic acid (Z-8b) [52] (Procedure A). ν (KBr/cm−1) 3150 - 2550 (bs, OH), 1678 (C=O), 1617 (C=C), 1432, 1289, 1210, 1073, 819, 713; δH (400 MHz, DMSO-d6) 2.11 (3H, d, 4J = 1.6 Hz, CH3), 5.92 (1H, q, 4J = 1.6 Hz), 7.25 (2H, d, 3J = 8.8 Hz), 7.39 (2H, d, 3J = 8.8 Hz); δH (400 MHz, DMSO-d6) 26.7 (CH3), 119.4 (CH), 128.2 (CH, 2C), 129.4 (CH, 2C), 132.6, 139.9, 152.6, 166.8 (Cquat, CO). (E)-3-(4- Chlorophenyl)but-2-enoic acid (E-8b). Beige solid, mp. 133˚C [Lit. 135˚C]; δH (400 MHz, DMSO-d6) 2.47 (3H, d, 4J = 1.6 Hz, CH3), 6.12 (1H, q, 4J = 1.6 Hz), 7.47 (2H, d, 3J = 8.8 Hz), 7.59 (2H, d, 3J = 8.8 Hz), 12.2 (1H, bs, OH); δC (100.5 MHz, DMSO-d6) 17.5 (CH3), 118.6 (CH), 128.5 (CH, 2C), 129.0 (CH, 2C), 134.2, 140.7, 152.6, 167.8 (CO, Cquat).
(E)-3-(4-Bromophenyl)but-2-enoic acid (8c) (Procedure A). Colorless solid, mp. 116˚C [Lit. 116˚C [11] ]; ν (KBr/cm−1) 3150 - 2550 (bs, OH), 1681 (C=O), 1610 (C=C), 817; δH (400 MHz, DMSO-d6) 2.47 (3H, q, 4J = 1.2 Hz, CH3), 6.12 (1H, d, 4J = 1.2 Hz), 7.51 (2H, d, 3J = 8.4 Hz), 7.60 (2H, d, 3J = 8.4 Hz), 12.30 (1H, bs, OH); δC (100.5 MHz, DMSO-d6) 17.5, 118.6, 122.9, 128.8 (2C), 131.9 (2C), 141.0, 152.7, 167.8.
(E)-3-(4-Iodophenyl)but-2-enoic acid (8d) (Procedure A) [53] . Colorless solid; mp. 161˚C; ν (KBr/cm−1) 3150 - 2550 (bs, OH), 1683 (C=O), 1608 (C=C), 1436, 1278 (C-O), 1208 (C-O), 994, 814, 708; δH (400 MHz, DMSO-d6) 2.45 (3H, q, 4J = 1.2 Hz, CH3), 6.11 (1H, q, 4J = 1.2 Hz), δH (400 MHz, CDCl3) 2.58 (3H, q, 4J = 1.2 Hz, CH3), 6.15 (1H, q, 4J = 1.2 Hz), 7.36 (2H, d, 3J = 8.4 Hz), 7.43 (2H, d, 3J = 8.4 Hz); δC (100.5 MHz, DMSO-d6) 17.4, 96.1, 118.4, 128.8 (2C), 137.8 (2C), 141.4, 152.9, 167.8; δC (100.5 MHz, CDCl3) 18.2 (CH3), 116.5 (CH), 127.7 (CH, 2C), 128.9 (CH, 2C), 135.4 (Cquat), 140.3 (Cquat), 157.1 (Cquat), 171.6 (Cquat, CO), and 3-(4-iodophenyl)but-3-enoic acid (8e) as a colorless solid, mp. 143˚C; ν (KBr/cm−1) 3150 - 2550 (bs, OH), 1685 (C=O); δH (400 MHz, CDCl3) 3.51 (2H, s), 5.28 (1H, s), 5.57 (1H, s), 7.17 (2H, d, 3J = 6.4 Hz), 7.65 (2H, d, 3J = 6.4 Hz); δC (100.5 MHz, CDCl3) 40.4 (CH2), 117.5 (CH2), 127.6 (2C, CH), 133.1 (Cquat), 137.6 (2C, CH), 138.9 (Cquat), 139.4 (Cquat), 174.9 (Cquat, CO).
(E)-3-Phenylpent-2-enoic acid (E-8f) (Procedure A). Colorless solid, mp. 93˚C [Lit. 95˚C [54] ]; ν (KBr/cm−1) 3150 - 2550 (bs, OH), 1698 (C=O); δH (400 MHz, CDCl3) 1.09 (3H, t, 3J = 7.6 Hz, CH3), 3.13 (2H, q, 3J = 7.6 Hz), 6.06 (1H, s), 7.38 - 7.40 (3H, m), 7.46 - 7.47 (2H, m), 11.7 (1H, s, OH); δC (100.5 MHz, CDCl3) 13.6 (CH3), 24.5 (CH2), 115.8 (CH), 126.7 (CH, 2C), 128.6 (CH, 2C), 129.2 (CH), 140.9 (Cquat), 165.0 (Cquat), 171.2 (Cquat); and (Z)-3-phenylpent-2-enoic acid (Z-8f) as a colorless solid, mp. 74 oC; ν(KBr/cm−1) 3180 - 2550 (bs, OH), 1687 (C=O); δH (400 MHz, CDCl3) 1.04 (3H, t, 3J = 7.2 Hz, CH3), 2.44 (2H, q, 3J = 7.2 Hz), 5.86 (1H, s), 7.13 - 7.15 (2H, m), 7.30 - 7.35 (3H, m); δC (100.5 MHz, CDCl3) 12.0 (CH3), 33.8 (CH2), 115.3 (CH), 127.0 (CH, 2C), 127.8 (CH), 128.0 (CH, 2C), 139.8 (Cquat), 163.8 (Cquat), 170.0 (Cquat).
(E)-4-Phenylbut-2-enoic acid (8g) (procedure A). Beige solid, mp. 96˚C [Lit. 97˚C [55] ]; νmax (KBr/cm−1) 3054, 1702 (C=O), 1409, 1218 (C-O), 977, 914, 745, 692; δH (400 MHz, DMSO-d6) 3.23 (2H, d, 3J = 7.2 Hz), 6.28 (1H, dd, 3J = 16.4 Hz, 3J = 7.2 Hz), 6.46 (1H, d, 3J = 16.4 Hz), 7.20 (1H, m), 7.28 (2H, dd, 3J = 7.2 Hz, 3J = 7.2 Hz), 7.38 (2H, d, 3J = 7.2 Hz); δH (400 MHz, CDCl3) 3.30 (2H, d, 3J = 8.4 Hz), 6.29 (1H, dd, 3J = 15.6 Hz, 3J = 8.4 Hz), 6.52 (1H, d, 3J = 15.6 Hz), 7.25 (1H, m), 7.32 (2H, m), 7.39 (2H, m); δC (100.5 MHz, DMSO-d6) 38.3 (CH2), 123.6 (CH), 126.4 (2C, CH), 127.9 (CH), 129.1 (2C, CH), 132.7 (CH), 137.1 (Cquat), 173.2 (CO); δC (100.5 MHz, CDCl3) 38.1 (CH2), 120.9 (CH), 126.3 (2C, CH), 127.7 CH), 128.6 (2C, CH), 134.0 (CH), 136.6 (Cquat), 177.8 (Cquat, CO).
Ethyl (E)-3-anthran-1-ylbut-2-enoate (10b). A mixture of 1-acetylanthracene (300 mg, 1.36 mmol), phosphorane 2a (760 mg, 2.18 mmol), and benzoic acid (30 mg, 0.25 mmol) in benzene (8 mL) was heated at 80˚C for 12 h. After 3 h, additional 2a (300 mg, 0.87 mmol) was added. The cooled reaction mixture was concentrated in vacuo and subjected to column chromatography on silica gel (ether/CHCl3/hexane 1:3:3) to give 10b (25 mg, 6%) as a colorless oil. ν (neat/cm−1) 1720 (C=O), 1634, 1170; δH (400 MHz, CDCl3) 1.35 (3H, t, 3J = 7.2 Hz), 2.70 (3H, d, 4J = 1.2 Hz), 4.28 (2H, q, 3J = 7.2 Hz), 6.07 (1H, q, 4J = 1.2 Hz), 7.25 (1H, d, 3J = 6.8 Hz), 7.42 (1H, dd, 3J = 8.4 Hz, 3J = 6.8 Hz), 7.47 - 7.48 (1H, m), 7.95 ? 8.00 (3H, m), 8.41 (1H, s), 8.44 (1H, s); δC (100.5 MHz, CDCl3) 14.3 (CH3), 21.6 (CH3), 60.0 (OCH2), 120.6 (CH), 123.5 (CH), 124.2 (CH), 124.6 (CH), 125.6 (CH), 125.8 (CH), 126.8 (CH), 127.3 (Cquat), 127.6 (Cquat), 127.9 (CH), 128.5 (2C, CH), 128.7 (Cquat), 131.5 (Cquat), 131.8 (Cquat), 142.1 (Cquat), 166.8 (Cquat, CO).
For the preparation of 12a - 12c, general procedure A was used. After acidification of the reaction medium after completion of the reaction, the aqueous phase was extracted with CHCl3. The organic phase was dried over MgSO4, concentrated in vacuo and the residue was filtered over a small layer of silica gel (ether/CHCl3 1:1).
5,9-Dimethyldeca-2,8-dienoic acid (12a) [56] as a colorless oil; νmax (neat/cm−1) 3100 - 2550 (bs, OH), 2917, 1695 (C=O), 1648, 1428, 1282, 983, 938, 686; δH (3H, d, 3J = 6.8 Hz), 0.95 (1H, m), 1.10 (1H, m), 1.35 (1H, m), 1.59 (3H, d, 4J = 0.8 Hz), 1.68 (3H, d, 4J = 0.8 Hz), 1.90 ? 2.07 (2H, m), 2.22 - 2.24 (2H, m), 5.04 (1H, m), 5.82 (1H, d, 3J = 15.6 Hz), 7.07 (1H, m); δC (100.5 MHz, CDCl3) 17.7, 19.5, 32.0, 25.5, 25.9, 36.7, 39.7, 121.8, 124.3, 131.6, 151.3, 171.9 (CO).
(E)-Dec-2-enoic acid (12b) [57] . Colorless oil; νmax (neat/cm−1) 1698 (C=O), 1660, 1422, 1310, 1300, 980, 940; δH (400 MHz, CDCl3) 0.83 (3H, t, 3J = 5.6 Hz, CH3), 1.22 (m), 1.41 (2H, m), 2.17 (2H, m), 5.76 (1H, d, 3J = 15.2 Hz), 7.03 (1H, dt, 3J = 15.2 Hz, 3J = 5.6 Hz); δC (100.5 MHz, CDCl3) 14.0 (CH3), 22.6 (CH2), 27.8 (CH2), 29.0 (CH2), 29.1 (CH2), 31.7 (CH2), 32.3 (CH2), 120.6 (CH), 152.4 (CH), 172.1 (Cquat, CO); MS (EI, 70 eV) m/z (%) 170 (M+, 13), 73 (65).
3-Cyclohexylpropenoic acid (12c). Colorless solid, mp. 53˚C [Lit. 57˚C - 58˚C [58] ], νmax (neat) 3400 - 2550 (bs, OH), 2931, 2856, 1699 (C=O); δH (400 MHz, CDCl3) 1.00 - 2.05 (10H, m), 2.20 (1H, m), 5.69 (1H, dd, 3J = 16.0 Hz, 4J = 2.0 Hz), 6.95 (1H, dd, 3J = 16.0 Hz, 3J = 6.8 Hz), 9.40 - 10.00 (1H, bs, OH); δC (100.5 MHz, CDCl3) 25.6, 25.9, 31.6, 40.5, 118.5, 158.2, 172.9.
2.3. General Procedure B
Phenylpropiolic acid (13b). To a mixture of benzaldehyde (1f, 763 mg, 7.2 mmol) and ethoxycarbonylbromomethylidenetriphenylphosphorane (2b, 4.0 g, 9.4 mmol) was given aq. NaOH solution (10 w%, 1.8 g NaOH in 18 mL H2O) and the resulting suspension was stirred at 85˚C for 16 h. Triphenylphosphine oxide was filtered off the cooled solution. Thereafter, the filtrate was acidified carefully with 15 w% aq. HCl. The resulting suspension was cooled and filtered. The solid obtained was dried in air to give phenylpropiolic acid (13b, 589 mg, 56%) as a colorless solid. An analytical sample gave mp. 133˚C [Lit. 135˚C - 137˚C [41] ]-νmax (KBr/cm−1) 3200 - 2500 (bs, OH), 2238, 2201 (C≡C), 1670 (C=O), 1489, 1417, 1305, 1208 (C-O), 919, 753, 683, 610, 533, 509; δH (400 MHz, CDCl3) 7.39 - 7.42 (2H, m), 7.46 - 7.48 (1H, m), 7.60 - 7.62 (2H, m), 10.1 (bs, OH); δC (100.5 MHz, CDCl3) 80.2 (Cquat), 88.8 (Cquat), 119.1 (Cquat), 128.8 (2C, CH), 131.1 (CH), 133.3 (2C, CH), 158.4 (Cquat, CO); MS (EI, 70 eV) m/z (%) 146 (M+) (100), 76 (30).
4-Methoxyphenylpropiolic acid (13a) (Procedure B). Pale yellow solid, mp. 143˚C [Lit. 144˚C [59] ]; νmax (KBr/cm−1) 2197 (C≡C), 1671 (C=O), 1602 (C=C), 1511, 1417, 1322, 1297, 1255, 1213, 1168, 1028, 923, 832, 799, 609, 565, 539; δH (400 MHz, CDCl3) 3.84 (3H, s, OCH3), 6.89 (2H, d, 3J = 8.8 Hz), 7.56 (2H, d, 3J = 8.8 Hz); δC (100.5 MHz, CDCl3) 55.5 (OCH3), 79.6 (Cquat), 90.0 (Cquat), 110.8 (Cquat), 114.4 (2C, CH), 135.3 (2C, CH), 158.4 (Cquat), 161.9 (Cquat, CO); δC (100.5 MHz, DMSO-d6) 55.8 (OCH3), 81.5 (Cquat), 85.9 (Cquat), 110.9 (Cquat), 115.1 (2C, CH), 135.1 (2C, CH), 155.1 (Cquat), 161.6 (Cquat, CO).
3-Chlorophenylpropiolic acid (13c) (Procedure B). Colorless solid, mp. 145˚C [Lit. 144˚C [60] ]; νmax (KBr/cm−1) 3200 - 2500 (bs, OH), 2960, 2586, 2217 (C≡C), 1683 (C=O), 1475, 1420, 1310, 1208, 877, 784, 674, 611, 519; δH (400 MHz, CDCl3); 9.0 (1H, bs, OH), 7.31 - 7.34 (1H, m), 7.44 - 7.49 (2H, m), 7.58 (1H, s); δC (100.5 MHz, DMSO-d6) 81.0 (Cquat), 86.3 (Cquat), 121.0 (Cquat), 129.9 (CH), 131.3 (2C, CH), 132.8 (CH), 134.5 (Cquat), 157.4 (Cquat).
3,4-Dimethoxyphenylpropiolic acid (13d) (Procedure B). Colorless solid, mp. 154˚C [Lit. 152˚C - 153˚C [61] ]; νmax (KBr/cm−1) 3200 - 2500 (bs, OH), 2937, 2837, 2595, 2202 (C≡C), 1684 (C=O), 1625, 1596, 1517, 1312, 1252, 1165, 1140, 1022, 975, 839, 810, 723, 579, 540. δH (400 MHz, CDCl3) 3.89 (3H, s, OCH3), 3.92 (3H, s, OCH3), δC (100.5 MHz, CDCl3) 79.4 (Cquat), 89.8 (Cquat), 110.9 (Cquat), 111.0 (CH), 115.4 (CH), 127.7 (CH), 148.8 (Cquat), 151.9 (Cquat), 157.9 (Cquat).
3. Results and Discussion
When a mixture of benzaldehyde 1 and ethoxymethylidenetriphenylphosphorane (2a, 1.3 eq.) is heated in a 10 w% aq. NaOH solution, sodium cinnamates are formed by Wittig olefination and subsequent ester hydrolysis. Upon neutralization of the aqueous solutions with 10 w% - 15w% aq. HCl, cinnamic acids 3 are obtained by easy filtration and drying of the solids in air. With this procedure, cinnamic acids 3 could be synthesized (Table 1).
Even solid 3e can be filtered off from the neutralized solution easily, as the anilinic nitrogen in 3e is not basic enough to be protonated upon careful neutralization of the reaction mixture. Most of the cinnamic acids are produced in good E-selectivity. However, for phenylpenta-2,5-dienoic acid (3p), 2,4-dimethoxycinnamic acid (3g), 2,5-dimethoxycinnamic acid (3h) and especially 3-bromo-2,5-dimethoxycinnamic acid (3i) anoticable quantity of Z-isomer is formed.
4-Nitrobenzaldehyde (1w) is not an adequate substrate for this one-pot procedure, even though 4-nitroben- zaldehyde (1w) and ethoxymethylidenetriphenylphosphorane (2a) give the Wittig product3wfacilely, when the reaction is performed in chloroform (Scheme 1). Also, the ester hydrolysis of 3w in 10 w% aq. NaOH proceeds
![]()
Table 1. Substituted cinnamic acids via one pot Wittig olefination-hydrolysis reaction.
*no additive; **tetramethylammonium bromide (0.15 eq. Bu4NBr) addad.
readily, and 3x is obtained, which can be filtered off and dried after acidification of the reaction solution. Yet, in 10 w% aq. NaOH, 4-nitrobenzaldehyde (1w) undergoes a rapid Cannizzaro reaction, which would be expected to be the general competing pathway for benzaldehydes in aqueous basic media. However, with the exception of the nitro-substituted benzaldehydes, Cannizzaro is not noted to be a main side reaction for the benzaldehydes used. The reason for this may be that the Wittig olefination ensues rapidly in the lipotropic droplets formed by themixture of benzaldehyde and phosphorane, initially suspended in the aqueous solution. In the case of 4-nitrobenzaldehyde (1r) in 10 w% aq. NaOH, a deep red homogeneous solution is produced immediately, from which one of the Cannizzaro products, 4-nitrobenzoic acid (4), can be obtained facilely by extraction of the side product with CH2Cl2 and acidification of the aqueous phase with subsequent extraction of the phase and crystallization of the obtained product [62] [63] . Cannizaro reaction can also be noted for 2-nitrobenzaldehyde under the conditions, and the 2-nitrobenzoic acid can be obtained easily.
In the case of 1x (Figure 1), only (Z)-p-benzyloxycinnamic acid (3y) was obtained as the cinnamic acid
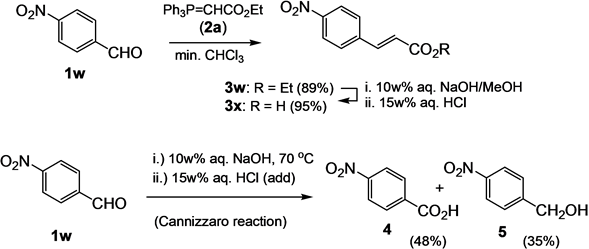
Scheme 1. Comparison of the sequential Wittig olefination?hydrolysis reaction of 4-nitrobenzaldehyde (1w) leading to cinnamic acid 3x and the reaction under one pot conditions leading to Cannizzaro products 4 and 5.
![]()
Figure 1. Reduction of alkenes carrying O-benzyl esterand/or O-benzyl ether func- tions.
fraction. The E-isomer could not be detected. At 70˚C, triphenylphosphine oxide and the Wittig product develop liquid hydrophobic micelles. Ester hydrolysis takes place at the micelle boundary, and it may be that the ethyl (E)- p-benzyloxycinnamate, more easily forming intermolecular layers within the micelle, is not as readily accessible as the Z-isomer. It must also be noticed that under the strongly basic conditions, the benzyloxy function is not sufficiently stable and is severed to provide the hydroxy-substituted compound. That micelles play a role in the ester hydrolysis can be seenin alkyl cinnamates with larger residues, such as in ethyl phenylpenta-2,4-dienoate, where the addition of tetramethylammonium bromide [(CH3)4NBr] leads to more yield in phenylpenta-2,4-dienoic acid (3p).
In cases, where the arylcarbaldehydes undergo Wittig reaction with alkoxycarbonylmethylidene phosphoranes slowly and only at higher temperatures at which the phosphoranes are already hydrolysed, a variant of the reaction protocol can be employed. First, the aldehyde is reacted with the phosphorane under solventless conditions at higher temperatures. Thereafter, 10 w% aq. NaOH is added to the reaction mixture, and the reaction is completed. 9-Anthranylcarbaldehyde (1t) represents such a case, where the aldehyde undergoes Wittig olefination only sluggishly due to the steric congestion around the carbaldehyde function. 1t can be reacted with 2a under solventless conditions at 130˚C [64] . Subsequent addition of aq. NaOH to the reaction and heating of the mixture gives the 3-(anthran-9-yl)propenoic acid (3z) in 85% yield (Scheme 2). It must be noted that upon cooling of the reaction, sodium 3-(anthran-9-yl)propenoate (3z-Na) crystallizes in the aqueous solution, and the resulting solid, which is comprised of the sodium salt of the acid as well as triphenylphosphine oxide, must be washed diligently with hot water upon filtration of the triphenylphosphine oxide. The precipitation was also noted in the hydrolysis of isolated ethyl anthranylpropenoate (3z-Et, R = Et) in 10 w% aq. NaOH. In that case, the precipitate can be treated directly with aq. HCl to yield anthranylpropenoic acid (3z) as a filterable precipitate.
The same solventless procedure was followed, when reacting 4-hydroxybenzaldehyde (1z) with phosphorane 2a at 140˚C for 90 min. Thereafter, the mixture was taken up in 10 w% aq. NaOH and stirred at rt for 30 min., after which the triphenylphosphine oxide was filtered off, and the filtrate acidified with half-conc. aq. HCl to give 4-hydroxycinnamic acid (6, p-coumaric acid) (Scheme 3). Previously, 4-hydroxycinnamic acid had been prepared by Horner Emmons olefination [65] with a subsequent, second hydrolysis step and via Wittig reaction with a polymer-bound Wittig reagent and a subsequent hydrolysis step [66] .
Acetophenones 7 have been subjected successfully to the one-pot Wittig olefination-hydrolysis, too (Table 2). In these cases, a mixture of phosphorane 2a and acetophenone 7 was heated, and thereafter an aq. NaOH solution was added. A workup identical to that described for the benzaldehydes provided the products as mixtures of

Scheme 2. Solventless Wittig reactionand subsequent hydrolysis without work-up of 3z.

Scheme 3. Preparation of p-coumaric acid (6) by solventless Wittig-olefination with basic extractive work-up.
![]()
Table 2. One pot Wittig reaction and hydrolysis with acetophenone as starting material.
E/Z-isomers. The E-isomer could be obtained in pure form by recrystallization from 1:1 v/v CH2Cl2/hexane. In order to obtain both isomers in pure form, the mixture was subjected to chromatography on silica gel (hexane/EtOAc 2:1). The reaction is dependent on the steric bulkiness of the ketone, where bulkiness in the ketone leads to a sterically congested transition state in the Wittig olefination. Thus, 1-acetylanthracene (9) does not react under the conditions to the respective acid at all. 9 reacts with phosphorane 2a sluggishly, and produces butenoate 10b after 12 h in refluxing chloroform in the presence of benzoic acid as acid catalyst only in trace amounts. Under the conditions of the one-pot Wittig olefination?ester hydrolysis, much of the phosphorane 2a hydrolyses before undergoing Wittig olefination with 9 (Scheme 4).
Alkanals and alicyclic carbaldehydes 11 have also been reacted in a one-pot Wittig olefination-hydrolysis reaction. The yields of the corresponding unsaturated acids 12 were slightly lower than for aromatic aldehydes. Also, the obtained carboxylic acids could not be purified simply by crystallization through addition of hydrochloric acid to the reaction mixture. Rather, they have to be extracted after the acidification of the reaction mixture and subjected to column chromatography on silica gel (Scheme 5).
When a mixture of (substituted) benzaldehyde 1 and ethoxybromomethylidenetriphenylphosphorane (2b, 1.3
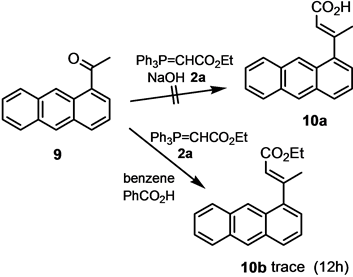
Scheme 4. Wittig reaction of 1-acetylanthracene (9) with phosphorane.
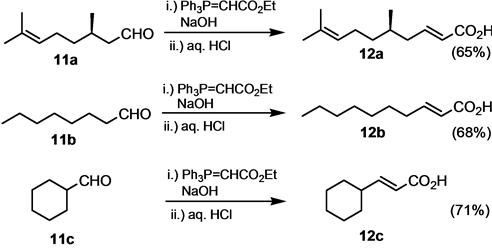
Scheme 5. One-pot transformation of alkanals and alicyclic aldehydes.
![]()
Table 3. Arylpropiolic acids from benzaldehydes in one pot synthesis in aq. solutions.
eq.) is heated in a 10 w% aq. NaOH solution, arylpropiolic acids 13 can be isolated upon careful neutralization of the aqueous solution (Table 3). In this case, the Wittig olefination is followed by ester hydrolysis and base catalysed dehydrobromination. Phenylpropiolic acids are usually prepared by bromination and dehydrobromination of cinnamic acids [60] or by Sonogashira coupling of haloarenes with propiolates [67] . In the present communication, we are showing a facile one-pot procedure to phenylpropiolic acids with a simple extractive work- up. For certain derivatives, cinnamic acids have been found as side-products. Nevertheless, the procedure in its yields compares well with that of Chenault and Dupin [59] , who employ ethoxyiodomethylidenetriphenylphosphorane in Na2CO3/methanol, followed by subsequent acid catalysed hydrolysis of the phenylpropiolates.
4. Conclusions
We have developed two simple procedures from benzaldehydes to cinnamic acids and to phenylpropiolic acids, respectively, utilizing a modified Wittig-olefination protocol. The reactions and the work-up are performed without any organic solvents in most cases here described. Yields are reproducibly good. The one-pot procedure of olefination and ester hydrolysis can be utilized with alkanals and acetophenones, also. Here, however, chromatographic separation of the products is necessary in some cases.
In former times, the Wittig olefination reaction has not been viewed as a green reaction due to its poor atom- economy. Recently, however, a dedicated effort has seen the development of numerous methods to recycle the toxic triphenylphosphine oxide [17] [68] - [70] that joins older methods [71] , and some of them already established in industrial processes [17] [72] . The reaction procedure described above allows for an efficient separation of triphenylphosphine oxide from the mixture upon completion of the reaction by simple filtration. Then, triphenylphosphine oxide is dried and stored and can be recycled upon accumulation from a larger number of Wittig reactions.
NOTES
![]()
*Corresponding author.