Received 24 November 2015; accepted 23 January 2016; published 26 January 2016

1. Introduction
Alzheimer’s disease (AD), one of the most common forms of dementia, is a progressive and neurodegenerative brain disorder that targets to cholinergic neurons of central nervous system (CNS) accompanied with spatial awareness and cognitive ability [1] . The risk of suffering this pathology increases sharply with age. However, the pathological beginning and advancement of AD are greatly complication.
Considering that the pathological beginning and advancement of AD are greatly complication and multi- factors effected, a single drug acting on a specific target to acquire the desired clinical effects might not be much effective [2] . Accordingly, the multitarget-directed ligand (MTDL) approach [3] has been increasingly investigated by many researchers, which have designed a lot of various compounds aiming to different targets by making use of this approach. In order to design effective dual inhibitors targeting to BACE 1 and AChE for the treatment of AD, the MTDL approach was also taken in this work.
As reported [4] , (−)-gallocatechin gallate (Figure 1, Figure 2) isolated from green tea, Camellia sinensis L., is a potent naturally occurring β-secretase (BACE 1) inhibitor. There are also several SAR studies [4] [5] analyz- ing the relationship between the structure of (−)-gallocatechin gallate and its inhibitory activity to BACE 1, which are greatly useful when starting a project to research novel BACE 1 inhibitors especially based the structure of the lead compound, (−)-gallocatechin gallate. From the SAR studies, the O-gallate moiety of (−)-gallocatechin gallate, which forms not strong interactions with BACE 1, was clipped from the original structure of (−)-gallo- catechin gallate when developing the scaffold in this work. On the contrary, the aromatic ring connected to C-2 of the structure of (−)-gallocatechin gallate is an important factor to BACE 1 inhibitory activity and therefore was kept. Meanwhile, (+)-(S)-dihydro-ar-tumerone (Figure 1, Figure 2) isolated from P. dasyrachis oil, was proved to be a potent AChE inhibitor possessing neuroprotective effects. And more interestingly, (+)-(S)-dihydro- ar-tumerone has a highly similar structure with (−)-gallocatechin gallate especially when they were superimposed together in a 3D model (Figure 1) by making use of Molecular Operating Environment (MOE 2008.10), which means that both BACE 1 and AChE inhibitors could have similar main structures at some degree.
![]()
Figure 1. The scaffolds of (+)-(s)-dihydro-ar-tuerone and (−)-gallo- catechin gallate (its’ gallate group was clipped) which were superim- posed together in a 3D model by using MOE 2008.10.
![]()
Figure 2. The design of 3 series of new derivates was based on the molecular skeleton, 6-amino-1,2-dihydroisoquinolin-3(4H)-one, which was generated from the 3D structures of compound (+)-(S)-dihydro- ar-tumerone and (−)-gallocatechin gallate by making use of Molecu- lar Operating Environment (MOE 2008.10). The substitutions R were acyl groups which were described in Table 1.
Then, a new molecular skeleton, tetrahydroisoquinolin-3-one fitting the common structure of the scaffolds of (+)-(S)-dihydro-ar-tumerone and (−)-gallocatechin gallate, was proposed, which aimed to design BACE 1 and AChE dual inhibitors by making use of Molecular Operating Environment (MOE 2008.10). At last, series of derivates based on this molecular skeleton was synthesized and its biological activity was evaluated.
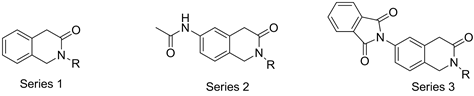
As depicted in Figure 2, the designed new molecules are divided into 3 series on the basis of the mainly structural differences between them. Firstly, there is an absence of nitrogen atom at 6-benzene position in the compounds of series 1. So, the contribution of nitrogen atom or the group connected nitrogen atom at 6-benzene position to enhance binding affinity will be revealed with such structural difference. Secondly, the existing of an aromatic group at the nitrogen atom of 6-benzene position in the structure of the compounds of series 3, especially comparing to the compounds in series 2, will help to understand whether the aromatic group can form π-π force with the residues of the pockets of enzymes. Lastly, there are also simple functional changes at N-2 in the structure of the compounds of series 1, 3 and 5. Noticeable, most of substitutions are aromatic groups; because the pharmacophore analysis indicated that an aromatic ring at these positions is essential to bind BACE 1.
2. Results and Discussion
2.1. Chemistry
The synthesis of the 3 series of new molecules (Scheme 1) had been processed by standard or reported methods, in very efficient and short synthetic sequences, with good overall yields. All new molecules gave satisfactory analytical and spectroscopic data, in good agreement with their structures.
2.2. AChE Inhibition
The AChE inhibitory activities of the new molecules were assayed using a modified version of the Ellman protocol in vitro [6] . In Table 1, the IC50 values of AChE inhibition were reported by using donepezil as control. All synthesized compounds possessed moderate to potent AChE inhibition (IC50 values ranging from 1 nM to 1000 nM) and several derivates had equipotent AChE inhibition with donepezil. From Table 1, it was obviously to find that there was an averagely inhibitory activity enhancement among 3 series (series 3 > series 2 > series 1). And the relevant docking studies (AchE, PDB ID: 4EY7) revealed that the N-phthaloyl in the compounds of series 3 can help them form a strong π-π force with the residues of the pocket which made the main contributions to their potent AChE inhibition (Figure 3).
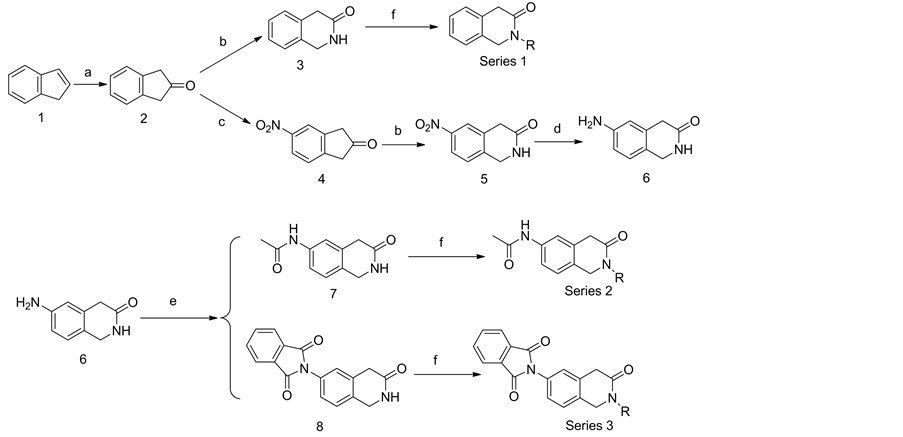
Scheme 1. Reagents and conditions: (a) HCO2H, H2O2, H2O, 2 h, 40˚C, 7 h, rt; (b) HCl, NaN3, H2O, 0˚C; 2 h, 0˚C; 0˚C to 25˚C; overnight, 25˚C; K2CO3, H2O, cooled, pH 9; (c) HNO3, −25˚C; (d) Zn, CH3COOH, THF; (e) AC2O, CH3COOH; (f) Various acyl chloride, anhydrous DMA, 0˚C - 100˚C and stirring for 4 h.
![]()
![]()
Table 1. AChE and BACE 1 inhibition activity for all synthesized compounds.
aThe in vitro test compound concentration required to produce 50% inhibition of AChE and BACE 1. The result (IC50) was the mean of three independent experiments.
![]()
![]()
Figure 3. Compound 3d docked with the pocket of AChE (a) the 3D model of this complex and (b) the force interactions between compound 3d and the residues of AChE in a 2D model.
2.3. β-Secretase Inhibition
The BACE 1 inhibitory activities of all synthesized compounds were tested using FRET (Fluorescence resonance energy transfer) method [7] based on the β-secretase Inhibitor Screening Assay Kit (Shanghai Haling biological technological CO., LTD., China). In Table 1, compounds containing a nitrogen atom at C-6 position of series 2 and 3 displayed more potency than those series not containing. Furthermore, compounds in series 3 also possessed more strongly inhibitory activities than those in series 1 and 2. Compound 3d was taken as an example of compounds in series 3 to do the docking studies (BACE 1, PDB ID: 1M4H), and the force interactions between 3d and the active sites of BACE 1 were described in 2D and 3D model (Figure 4). In Figure 4, compound 3d had an arene-cation interaction with Arg 128 which belongs to the S3’ sub-pocket of BACE 1 and can also form hydrogen bonds with Asp 32 (belongs to the catalytic sites), Arg 235 (belongs to the S2 sub-pocket) and Thr 72 (belongs to the S2 sub-pocket).
2.4. Hydrogen Peroxide Scavenging Activity
In order to further evaluate the antioxidant activity of the synthesized derivatives, compounds 1d, 2e, 2j, 3d and 3f were analyzed using the ferrous ion oxidation-xylenol orange (FOX) assay [8] . The results shown in Figure 5 demonstrate that 1d, 2e, 2j, 3d and 3f possess obvious scavenging activities for hydrogen peroxide at the slightly high concentrations, while the scavenging activities are not noticeable as the compounds concentration less than 100 μM. Meanwhile, the results indicate that 1d, 2e, 2j, 3d and 3f almost have the same hydrogen peroxide scavenging activity on different concentrations.
2.5. Cytotoxicity Assay
The CCK-8 assay [9] was taken in this work to test the cytotoxicity of compound 1d, 2e, 2j, 3d and 3f on HEK 293 cells. And compound 1d, 2e, 2j, 3d and 3f was tested in five different concentrations (1, 10, 50, 100, and 200 μM in final concentration) respectively. The values in Figure 6 demonstrate that all these tested compounds do not show obvious toxicity in the CCK-8 assay, suggesting that 1d, 2e, 2j, 3d and 3f lack general toxicity.
![]()
![]()
Figure 4. A representation of molecular docking derived binding pose (3D) of compound 3d in the active site of BACE-1 (a) and docking simulated binding mode (2D) of compound 3d bound in the active site of BACE-1 (b).
![]()
Figure 5. Scavenging effect of 1d, 2e, 2j, 3d and 3f on hydrogen peroxide, which determined by the FOX assay with slight change. The results reported are the mean ± SD of three independent experiments.
![]()
Figure 6. The cell viability of compound 1d, 2e, 2j, 3d and 3f on HEK 293 cells using CCK-8 assay kits. The results reported are the mean ± SD of three independent experiments.
3. Experimental Procedures
All chemical reagents in this work were commercially available and solvents were dried in standard methods before using. Melting points were determined on an X4 melting point apparatus (Beijing Tech Instrument CO., LTD., China) and were uncorrected. 1H NMR spectra were recorded on a BRUKER 500 MHz or 400 MHz spectrometer with TMS as the internal reference. Reactions were monitored by thin-layer chromatography (TLC) on Merck (0.25 mm) glass-packed precoated silica gel plates (60 F254) and column chromatography was performed on silica Gel 60 (230 mesh). The purity of the synthesized compounds was determined by HPLC system (HP 1100 series, Agilent Technologies, Palo Alto, USA) and their final purity was all above 95%. MS spectra were recorded on Waters ZQ 4000 apparatus (electrospray ionization, ESI).
3.1. Synthesis Section
The procedure for synthesis of 1H-inden-2(3H)-one (2) [10]
To a three-necked flask hydrogen peroxide (7 ml, 30%) and formic acid (35 ml, 88%) were added, then a dropping funnel, stirrer and thermometer were put on its necks. When thetemperature was stabilized at 40˚C, indene (compound 1, 11.7 ml) was added using dropping funnel, with stirring overa period of 3 h. The reaction mixture was kept stirring at 40˚C for another 7 h. A yellowishbrown crystalline solid was obtained after removing the excess formic acid underreduced pressure. To a tow-necked flask equipped with a condenser sulfuric acid (100 ml, 7%) was added, and after the solution was heated to boiling, the obtained solid was added over several times. The mixture was steamdistilled and the steam was cooled and gathered. The gathered solution was further cold at 10˚C over a night and then filtered, to generate white crystals. Compound 2 was obtained after the white crystals were further dried in a vacuum under room temperature over 24 h. The yield is 60% - 75%, mp 56˚C - 58˚C (lit [11] , 54˚C - 56˚C).
The procedure for synthesis of 1,2-dihydroisoquinolin-3(4H)-one (3) [12]
In an ice-cooled solution of Compound 2 (2.0 g, 15.1 mmol, 1.0 equiv) in concentrated HCl (50 mL), NaN3 (2.0 g, 30.1 mmol, 2.0 equiv) was added portionwise. The reaction mixture was kept stirring for 2 h at 0˚C, then slowly warmed to room temperature and kept stirring for a night. After the mixture was quenched with ice water (50 mL) and basified to pH 9 using K2CO3, the solution was extracted with CH2Cl2. Next, the organic layer was dried with anhydrous Na2SO4, filtered and evaporated, to give a crude yellow solid which was further purified by flash chromatography on silica gel (hexane/EtOAc: 1/1 to EtOAc). The purified compound 3 is a light yellow solid (1.45 g, 65% yield), mp 143˚C - 145˚C (lit [13] , 146˚C - 148˚C).
The procedure for synthesis of 5-nitro-1H-inden-2(3H)-one (4) [14]
To a cold (−25˚C) concentrated nitric acid (16 mL, 90%), Compound 2 (1.4 mg, 10.0 mmol) was added portionwise with vigorous stirring. The mixture was stirred for another 30 min at −25˚C. Then the mixture was removed to 0˚C and stirred for another 1 h. Next, the reaction mixture was poured into a mixture of sodium hydroxide (13.0 g) in water and ice (ca. 100 mL), and extracted with ethyl acetate. The combined organic layer was washed with water, dried with anhydrous Na2SO4 and evaporated under reduced pressure, to give a crude yellow solid. Then the crude product was further purified by flash chromatography on silica gel (chloroform). The purified compound 4 is a yellow solid (1.02 g, 54% yield), mp 138˚C - 140˚C (lit. [14] , 141˚C - 143˚C).
The procedure for synthesis of 6-nitro-1,2-dihydroisoquinolin-3(4H)-one (5)
Compund 5 was synthesized as the procedure for synthesis of Compound 3. Compund 5 is a yellow solid, m.p: 159˚C - 161˚C, ESI-MS (m/z): 193 (M + H+), 1H NMR (400 MHz, CDCl3) δ 8.19 - 8.11 (m, 2H), 7.38 (d, J = 8.4 Hz, 1H), 6.70 (s, 1H), 4.62 (s, 2H), 3.71 (s, 2H).
The procedure for synthesis of 6-amino-1,2-dihydroisoquinolin-3(4H)-one (6) [15]
In a solution of Zn (70 mmol) aerated with ethylic acid (3 mL, 50 mmol) in THF (40 mL), Compound 5 (2.0 g, 3.3 mmol) was added with vigorous stirring. The reaction mixture was kept stirring for 12 h, under the protection of N2 at 65˚C. Then the mixture was filtered and the solvent was removed under reduced pressure. Next, the obtained residue was further washed by 5% NaOH (30 mL) to generate a crude brown solid. Then the crude product was further purified by flash chromatography on silica gel (Ethyl acetate/glacial acetic acid, 150:2). The purified compound 6 is a brown solid (1.40 g, 83% yield), mp 153˚C - 155˚C. ESI-MS (m/z): 163 (M + H+), 1H NMR (500 MHz, DMSO) δ 7.89 (d, J = 19.9 Hz, 1H), 6.83 (dd, J = 29.7, 8.0 Hz, 1H), 6.45 - 6.37 (m, 1H), 6.33 (s, 1H), 4.89 (d, J = 11.3 Hz, 2H), 4.16 (s, 2H), 3.25 (d, J = 15.8 Hz, 2H).
The general procedure for synthesis of N-(3-oxo-1,2,3,4-tetrahydroisoquinolin-6-yl)acetamide (7) [16]
Compound 6 (1.22 g, 7.5 mmol) was added to a solution of acetic anhydride (10 mL) and the reaction was kept at room temperature for 5 h. Then the mixture was poured into ice water. The precipitate was filtered, washed with water, and dried to give the compound 7 (1.27 g, 83%), m.p. 89˚C - 91˚C. ESI-MS (m/z): 205 (M + H+), 1H NMR (400 MHz, Chloroform-d) δ 7.48 (d, J = 8.4 Hz, 2H), 7.39 (s, 1H), 7.26 (s, 1H), 6.46 (s, 1H), 4.73 (s, 2H), 3.79 (d, J = 5.8 Hz, 2H), 2.19 (s, 3H).
The general procedure for synthesis of 2-(3-oxo-1,2,3,4-tetrahydroisoquinolin-6-yl)isoindoline-1,3-dione (8) [16]
Compound 6 (1.22 g, 7.5 mmol) was added to a solution of phthalic anhydride (2.25 g, 15.0 mmol) in acetic acid (25 ml) and the reaction was kept at 100˚C for 1 h. Then the mixture was poured into ice water. The precipitate was filtered, washed with water, and dried to give the compound 8 (1.91 g, 87%), m.p. 207˚C - 209˚C. ESI-MS (m/z): 293 (M + H+), 1H NMR (400 MHz, Chloroform-d) δ 7.99 (dd, J = 5.5, 2.9 Hz, 2H), 7.83 (dd, J = 5.5, 3.0 Hz, 2H), 7.47 (s, 3H), 6.47 (s, 1H), 4.76 (s, 2H), 3.60 (d, J = 5.9 Hz, 2H).
The general procedure for synthesis of series 1, 2 and 3 [17]
To a solution of compound 3 (compound 7 or compound 8, 1.0 mmol) in anhydrous dioxane (5 ml), 4-dime- thylaminepyridine (10 mg) and anhydrous potassium carbonate (100 mg) was added at room temperature. Then different kinds of acyl chloride (1.5 mmol) was added respectively and the mixture was kept stirring at reflux for 0.5 to 3 h. Next, after the solution was cooled to 0˚C, HCl (5%) was added to change the solution to pH 1 and extracted by chloroform. The organic layer was dried with anhydrous Na2SO4, filtered and evaporated, to give the relevant crude product which was further purified by flash chromatography on silica gel.
2-acetyl-1,2-dihydroisoquinolin-3(4H)-one (1b)
Compound 1b was found to be yellowish oil, yield 43%. ESI-MS (m/z): 190 (M + H+). 1H NMR (400 MHz, Chloroform-d) δ 7.34 (t, J = 6.1 Hz, 2H), 7.29 (d, J = 6.8 Hz, 2H), 4.60 (d, J = 3.6 Hz, 2H), 3.83 - 3.76 (m, 2H), 2.13 (d, J = 2.1 Hz, 3H).
2-benzoyl-1,2-dihydroisoquinolin-3(4H)-one (1c)
Compound 1c was found to be orange oil, yield 45%. ESI-MS (m/z): 252 (M + H+). 1H NMR (400 MHz, Chloroform-d) δ 7.30 (d, J = 7.2 Hz, 2H), 7.22 (s, 5H), 7.19 - 7.13 (m, 2H), 4.62 (s, 2H), 3.72 (d, J = 5.8 Hz, 2H).
2-(3-methylbenzoyl)-1,2-dihydroisoquinolin-3(4H)-one (1d)
Compound 1d was found to be sandy-brown oil, yield 47%. ESI-MS (m/z): 266 (M + H+). 1H NMR (400 MHz, DMSO-d6) δ 7.84 (d, J = 7.8 Hz, 2H), 7.43 (t, J = 7.7 Hz, 1H), 7.36 ? 7.30 (m, 2H), 7.25 (dd, J = 15.4, 6.9 Hz, 3H), 4.78 (s, 2H), 3.63 (d, J = 6.0 Hz, 2H), 2.39 (s, 3H).
2-(2-methoxybenzoyl)-1,2-dihydroisoquinolin-3(4H)-one (1f)
Compound 1f was found to be rufous oil, yield 42%. ESI-MS (m/z): 282 (M + H+). 1H NMR (400 MHz, Chloroform-d) δ 7.82 (dd, J = 7.7, 1.7 Hz, 1H), 7.50 - 7.43 (m, 1H), 7.34 (d, J = 6.5 Hz, 4H), 7.04 (t, J = 7.5 Hz, 1H), 6.92 (d, J = 8.4 Hz, 1H), 4.91 (s, 2H), 3.86 (d, J = 5.5 Hz, 2H), 3.43 (s, 3H).
2-(2-phenylacetyl)-1,2-dihydroisoquinolin-3(4H)-one (1g)
Compound 1g was found to be yellowish oil, yield 52%. ESI-MS (m/z): 266 (M + H+). 1H NMR (400 MHz, Chloroform-d) δ 7.32 (d, J = 7.2 Hz, 2H), 7.22 (s, 5H), 7.18 - 7.14 (m, 2H), 4.62 (s, 2H), 3.69 (s, 2H), 3.63 (d, J = 5.8 Hz, 2H).
N-(2-acetyl-3-oxo-1,2,3,4-tetrahydroisoquinolin-6-yl)acetamide (2b)
Compound 2b found to be yellowish oil, yield 43%. ESI-MS (m/z): 247 (M + H+). 1H NMR (400 MHz, DMSO-d6) δ 8.85 (s, 1H), 7.20 (d, J = 8.0 Hz, 2H), 7.09 (s, 1H), 4.25 (s, 2H), 3.38 (d, J = 5.8 Hz, 2H), 1.81 (d, J = 7.1 Hz, 6H).
N-(2-benzoyl-3-oxo-1,2,3,4-tetrahydroisoquinolin-6-yl)acetamide (2c)
Compound 2c was found to be orange solid, yield 45%. ESI-MS (m/z): 309 (M + H+), m.p. 151˚C - 153˚C. 1H NMR (400 MHz, Chloroform-d) δ 7.67 (s, 1H), 7.46 (d, J = 8.3 Hz, 2H), 7.26 (s, 5H), 7.11 (s, 1H), 4.63 (s, 2H), 3.55 (d, J = 5.8 Hz, 2H), 2.19 (s, 3H).
N-(2-(3-methylbenzoyl)-3-oxo-1,2,3,4-tetrahydroisoquinolin-6-yl)acetamide (2d)
Compound 2d was found to be yellow solid, yield 48%. ESI-MS (m/z): 323 (M + H+), m.p. 135˚C - 137˚C. 1H NMR (400 MHz, Chloroform-d) δ 7.87 (s, 1H), 7.48 (d, J = 6.7 Hz, 2H), 7.44 (d, J = 6.0 Hz, 1H), 7.37 (t, J = 6.1 Hz, 1H), 7.28 - 7.21 (m, 3H), 4.89 (s, 2H), 3.82 (d, J = 4.7 Hz, 2H), 2.43 (s, 3H), 2.19 (s, 3H).
N-(2-(3-chlorobenzoyl)-3-oxo-1,2,3,4-tetrahydroisoquinolin-6-yl)acetamide (2e)
Compound 2e was found to be yellow solid, yield 42%. ESI-MS (m/z): 343 (M + H+), m.p. 143˚C - 145˚C. 1H NMR (400 MHz, Chloroform-d) δ 7.51 - 7.43 (m, 2H), 7.31 - 7.29 (m, 1H), 7.27 (d, J = 2.3 Hz, 1H), 7.24 - 7.18 (m, 2H), 7.15 (t, J = 6.7 Hz, 2H), 4.64 (s, 2H), 3.66 (s, 2H), 2.29 - 2.09 (m, 3H).
N-(2-(2-methoxybenzoyl)-3-oxo-1,2,3,4-tetrahydroisoquinolin-6-yl)acetamide (2f)
Compound 2f was found to be light brown solid, yield 44%. ESI-MS (m/z): 339 (M + H+), m.p. 117˚C - 119˚C. 1H NMR (400 MHz, Chloroform-d) δ 8.10 (d, J = 7.4 Hz, 1H), 7.83 (dd, J = 7.7, 1.8 Hz, 1H), 7.51 (t, J = 7.9 Hz, 3H), 7.27 - 7.23 (m, 1H), 7.03 (t, J = 7.6 Hz, 1H), 6.93 (d, J = 8.4 Hz, 1H), 4.88 (s, 2H), 3.79 (d, J = 5.6 Hz, 2H), 3.51 (s, 3H), 2.16 (s, 3H).
N-(3-oxo-2-(2-phenylacetyl)-1,2,3,4-tetrahydroisoquinolin-6-yl)acetamide (2g)
Compound 4g was found to be yellowish oil, yield 48%. ESI-MS (m/z): 323 (M + H+). 1H NMR (400 MHz, Chloroform-d) δ 7.62 (s, 1H), 7.46 (d, J = 8.3 Hz, 2H), 7.26 (s, 5H), 7.11 (s, 1H), 4.63 (s, 2H), 3.72 (s, 2H), 3.55 (d, J = 5.8 Hz, 2H), 2.19 (s, 3H).
N-(3-oxo-2-(3-phenylpropanoyl)-1,2,3,4-tetrahydroisoquinolin-6-yl)acetamide (2j)
Compound 2j was found to be light brown oil, yield 52%. ESI-MS (m/z): 337 (M + H+). 1H NMR (400 MHz, Chloroform-d) δ 7.65 (s, 1H), 7.45 (d, J = 6.5 Hz, 2H), 7.28 - 7.23 (m, 2H), 7.20 (d, J = 5.1 Hz, 3H), 7.16 (s, 1H), 4.59 (s, 2H), 3.64 (d, J = 4.7 Hz, 2H), 2.99 (t, J = 5.9 Hz, 2H), 2.77 (t, J = 6.0 Hz, 2H), 2.17 (s, 3H).
N-(2-nicotinoyl-3-oxo-1,2,3,4-tetrahydroisoquinolin-6-yl)acetamide (2k)
Compound 2k found to be light red solid, yield 47%. ESI-MS (m/z): 310 (M + H+), m.p. 168˚C - 170˚C. 1H NMR (400 MHz, Chloroform-d) δ 9.24 (d, J = 2.1 Hz, 1H), 8.82 (dd, J = 4.9, 1.7 Hz, 1H), 8.33 (dt, J = 8.0, 1.9 Hz, 1H), 7.46 (d, J = 8.6 Hz, 3H), 7.40 (d, J = 8.4 Hz, 1H), 4.91 (s, 2H), 3.79 (d, J = 5.9 Hz, 2H), 2.17 (s, 3H).
2-(2-acetyl-3-oxo-1,2,3,4-tetrahydroisoquinolin-6-yl)isoindoline-1,3-dione (3b)
Compound 3b was found to be yellow solid, yield 45%. ESI-MS (m/z): 335 (M + H+), m.p. 135˚C - 137˚C. 1H NMR (400 MHz, Chloroform-d) δ 8.01 - 7.96 (m, 2H), 7.83 (dd, J = 4.4, 2.4 Hz, 2H), 7.48 - 7.44 (m, 3H), 4.65 (s, 2H), 3.59 (d, J = 4.6 Hz, 2H), 2.19 (d, J = 0.6 Hz, 3H).
2-(2-benzoyl-3-oxo-1,2,3,4-tetrahydroisoquinolin-6-yl)isoindoline-1,3-dione (3c)
Compound 3c was found to be yellow solid, yield 43%. ESI-MS (m/z): 397 (M + H+), m.p. 196˚C - 198˚C. 1H NMR (400 MHz, Chloroform-d) δ 8.08 (t, J = 1.5 Hz, 2H), 7.98 (dd, J = 5.5, 3.0 Hz, 2H), 7.83 (dd, J = 5.5, 3.1 Hz, 2H), 7.69 - 7.61 (m, 6H), 4.95 (s, 2H), 3.94 (d, J = 6.0 Hz, 2H).
2-(2-(3-methylbenzoyl)-3-oxo-1,2,3,4-tetrahydroisoquinolin-6-yl)isoindoline-1,3-dione (3d)
Compound 3d was found to be orange solid, yield 46%. ESI-MS (m/z): 411 (M + H+), m.p. 184˚C - 186˚C. 1H NMR (400 MHz, Chloroform-d) δ 7.98 (dd, J = 4.5, 2.5 Hz, 2H), 7.91 - 7.86 (m, 2H), 7.82 (dd, J = 4.4, 2.5 Hz, 2H), 7.46 (q, J = 7.8, 7.2 Hz, 5H), 4.92 (s, 2H), 3.93 (d, J = 4.8 Hz, 2H), 2.44 (s, 3H).
2-(2-(2-methoxybenzoyl)-3-oxo-1,2,3,4-tetrahydroisoquinolin-6-yl)isoindoline-1,3-dione (3f)
Compound 3f was found to be brown solid, yield 42%. ESI-MS (m/z): 427 (M + H+), m.p. 157-159℃. 1H NMR (400 MHz, Chloroform-d) δ 7.99 (dd, J = 5.5, 3.1 Hz, 2H), 7.84 (dd, J = 5.4, 3.1 Hz, 2H), 7.55 ? 7.50 (m, 3H), 7.48 - 7.44 (m, 3H), 7.06 (td, J = 7.5, 1.0 Hz, 1H), 4.93 (s, 2H), 3.93 (d, J = 5.5 Hz, 2H), 3.50 (s, 3H).
2-(3-oxo-2-(2-phenylacetyl)-1,2,3,4-tetrahydroisoquinolin-6-yl)isoindoline-1,3-dione (3g)
Compound 3g was found to be orange oil, yield 42%. ESI-MS (m/z): 411 (M + H+). 1H NMR (400 MHz, Chloroform-d) δ 8.00 (dd, J = 5.5, 3.0 Hz, 2H), 7.84 (dd, J = 5.5, 3.1 Hz, 2H), 7.45 (d, J = 8.5 Hz, 2H), 7.32 (d, J = 8.4 Hz, 2H), 7.27 (s, 4H), 4.67 (s, 2H), 3.74 (s, 2H), 3.61 (d, J = 5.8 Hz, 2H).
2-(2-(2-(4-methoxyphenyl)acetyl)-3-oxo-1,2,3,4-tetrahydroisoquinolin-6-yl)isoindoline-1,3-dione (3h)
Compound 3h was found to be claybank oil, yield 47%. ESI-MS (m/z): 441 (M + H+). 1H NMR (400 MHz, Chloroform-d) δ 7.98 (dd, J = 4.5, 2.4 Hz, 2H), 7.83 (ddd, J = 4.4, 2.3, 1.2 Hz, 2H), 7.36 - 7.31 (m, 2H), 7.22 - 7.17 (m, 2H), 6.90 - 6.86 (m, 1H), 6.85 - 6.81 (m, 2H), 4.68 - 4.62 (m, 2H), 3.82 - 3.78 (m, 2H), 3.77 - 3.73 (m, 3H), 3.42 (d, J = 4.6 Hz, 2H).
2-(3-oxo-2-(3-phenylpropanoyl)-1,2,3,4-tetrahydroisoquinolin-6-yl)isoindoline-1,3-dione (3j)
Compound 3j was found to be sundown oil, yield 43%. ESI-MS (m/z): 425 (M + H+). 1H NMR (400 MHz, Chloroform-d) δ 7.99 (dd, J = 5.5, 3.0 Hz, 1H), 7.83 (dd, J = 5.5, 3.0 Hz, 2H), 7.45 (d, J = 8.4 Hz, 2H), 7.40 (d, J = 8.4 Hz, 2H), 7.28 - 7.19 (m, 3H), 4.63 (s, 2H), 3.77 (d, J = 5.9 Hz, 2H), 3.02 (t, J = 7.5 Hz, 2H), 2.80 (t, J = 7.4 Hz, 2H).
2-(2-nicotinoyl-3-oxo-1,2,3,4-tetrahydroisoquinolin-6-yl)isoindoline-1,3-dione (3k)
Compound 6k was found to be reddish brown solid, yield 41%. ESI-MS (m/z): 398 (M + H+), m.p. 215˚C - 217˚C. 1H NMR (400 MHz, Chloroform-d) δ 9.29 (dd, J = 1.8, 0.7 Hz, 1H), 8.84 (dd, J = 3.9, 1.4 Hz, 1H), 8.36 (dt, J = 6.4, 1.6 Hz, 1H), 7.97 (dd, J = 4.4, 2.4 Hz, 2H), 7.82 (dd, J = 4.3, 2.4 Hz, 2H), 7.48 - 7.44 (m, 4H), 4.94 (s, 2H), 3.62 (d, J = 4.7 Hz, 2H).
3.2. Bio-Evaluation
In vitro AChE inhibitory activity screening [6]
The modified Ellman method was taken in this work to test the AChE inhibitory activity of the synthesized compounds. The enzyme AChE purchased from Sigma Chemical and the positive control donepezil purchased from Dalian Meilun Biotech (China). Meanwhile, 5,5-Dithio-bis(2-nitrobenzoic) acid (DTNB, 0.2 mM) was taken as color agent and acetylthiocholine (0.4 mM) as substrates. All compounds were determined in 100 mM phosphate buffer (pH 8.0) at 30˚C and IC50 values were calculated through UV spectroscopy based on the absorbance changes at 412 nm. Data are displayed as means±SD depending on three different experiments at least.
In vitro BACE 1 inhibit activity screening [7]
The FRET-based enzymatic kits (purchased from Shanghai Haling biological technological CO., LTD., China) were used in this work to test the BACE 1 inhibitory activities of all synthesized compounds. And the assay was carried out by following the manufacturers’ instructions. Meanwhile, the excitation and emission wavelength were set to 340 and 490 nm respectively to evaluate the hydrolysis of substrate. The final data are displayed as means ± SD depending on three different experiments at least.
Hydrogen peroxide scavenging activity assay [8]
Hydrogen peroxide scavenging activity was determined by a slightly modified version of ferrous ion oxidation-xylenol orange (FOX) assay. Firstly, to prepare the FOX reagent, nine fold volumes of reagent A, 4.4 mM butylated hydroxytoluene (BHT) in methanol, was added to one fold volume of reagent B, 2.56 mM ammonium ferrous sulfate and 1 mM xylenol orange in 250 µM H2SO4. Secondly, 400 µL of the test compounds with different concentrations were added to 100 μL of 1 mM H2O2 and incubated for 10 h at 37˚C in the dark. And then, 500 µL of the FOX agent was added. Thirdly, the reaction mixture was shaken and incubated for 30 min at room temperature. The appearing of a violet color in the back of the addition of the FOX agent presents a positive control reaction. On the contrary, discoloration is linked to scavenging activity. Lastly, to determine the absorbance of the mixture, the complex was evaluated at 560 nm. Notable, the FOX reagent with H2O2 acted as a control, while the FOX reagent lacking of compound and H2O2 acted as a blank.
Cytotoxicity assay [9]
The Human Embryonic Kidney 293 cells (HEK293) were taken in this test and cultured with the DMEM medium in a humidified environment at 37˚C with 5% CO2. The media was supplemented with 100 units/ml penicillin, 10% fetal bovine serum (FBS) and 100 units/ml streptomycin. Cells were sub-cultured and grown in collagen-coated tissue culture flasks. The cells were taken for the assay when they become 70% confluent. The compound cytotoxicity was reflected by measuring the cells’ ability to reduce WST-8 to WST-8 formazan. The cells were seeded into 96-well culture plate with proper concentration (5 × 103) at their exponential growth phase, then incubated in a CO2 incubator overnight. Next, 5 µL compounds (1, 10, 50, 100 and 200 µM in final concentration) were added to each well of the plate and incubated for another 4 h. Then, 10 µL WSK-8 (at final concentration 0.3 mg/ml in DMEM without phenol red) was added to each well and incubated for another 2 h. At last, the absorbance values of each well were obtained by a microplate reader at 450 nm. Cells without the test compounds acted as positive control and its assay values were set to 100%. The test compounds were dissolved in DMSO at 10 mM concentration firstly and then diluted to above-mentioned concentration in 10 mM phosphate buffer pH 7.4. Data of this test was recorded depending on three independent experiments and values of each well were read in triplicate.
4. Conclusion
Acknowledgements
The financial support from the National Natural Science Foundation of China (No. 20876180) is gratefully acknowledged. The authors also would like to thank School of Pharmaceutical Sciences of Central South University for support of MOE and microplate reader for this research project.
NOTES
![]()
*Corresponding author.